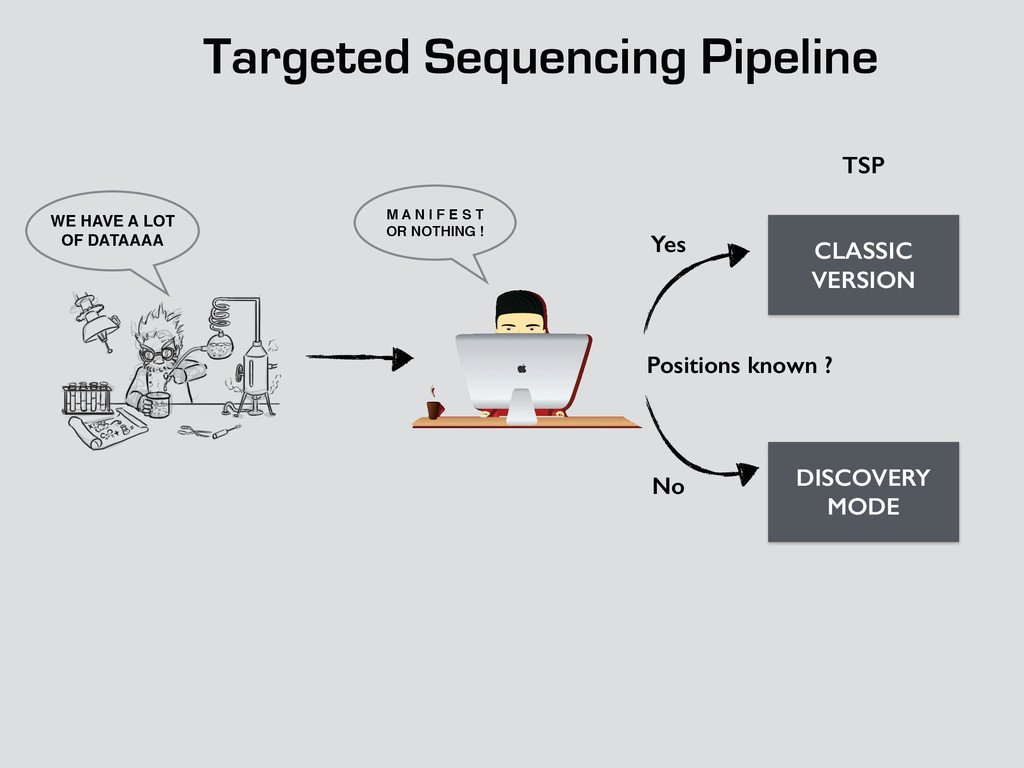
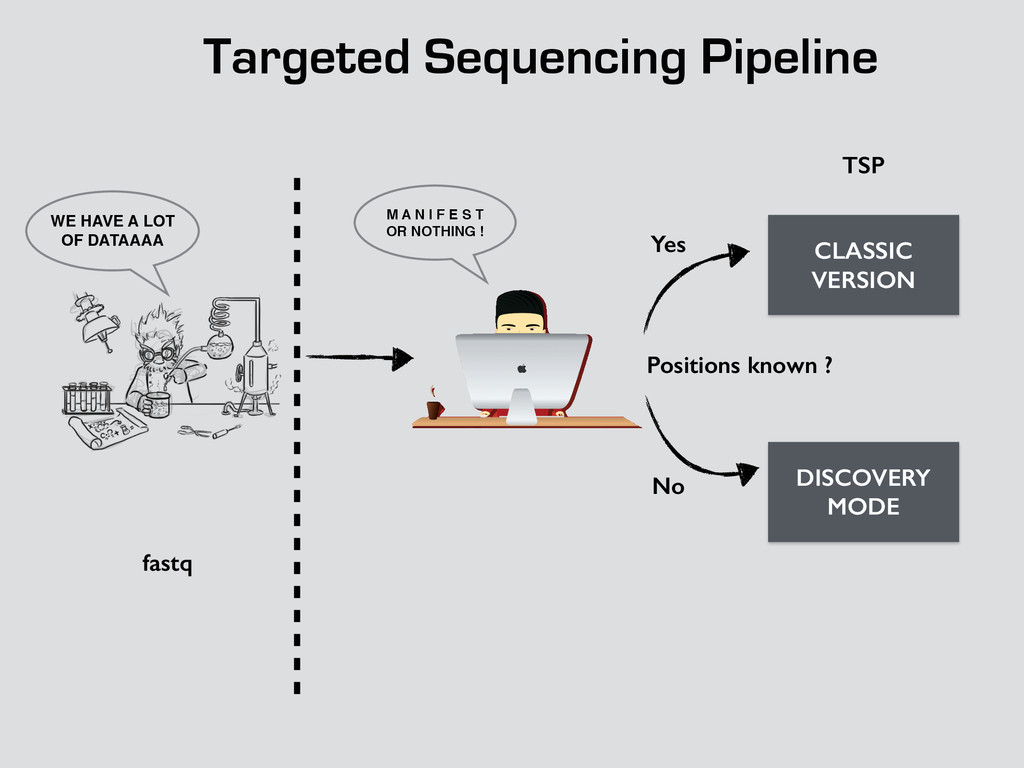
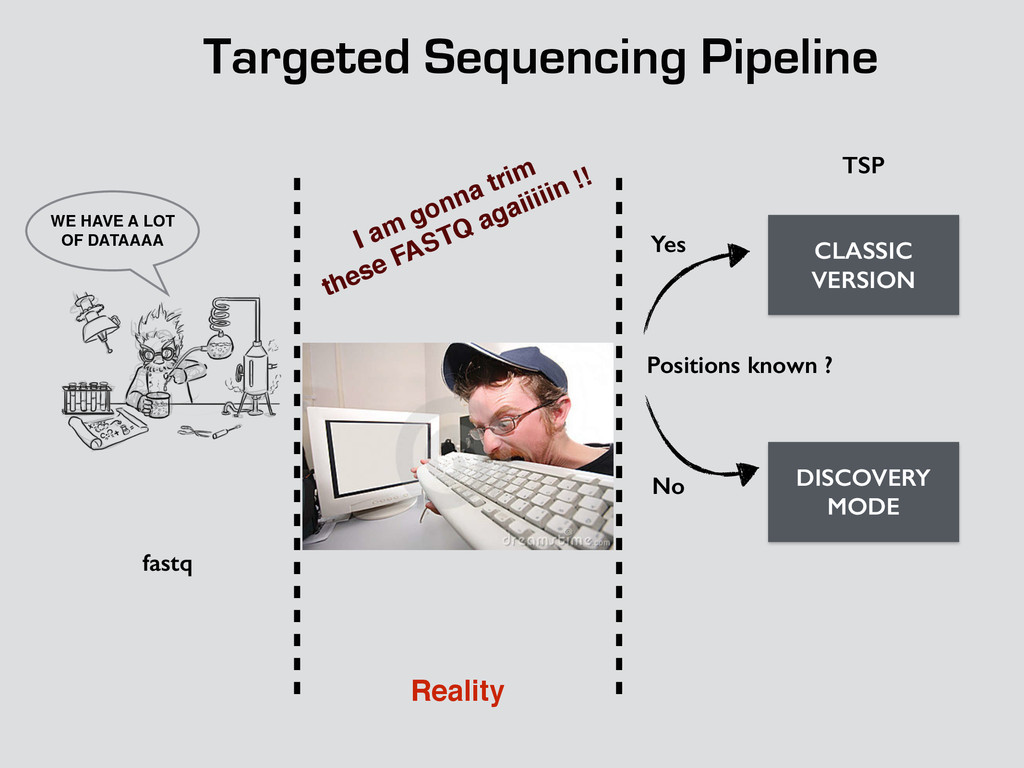
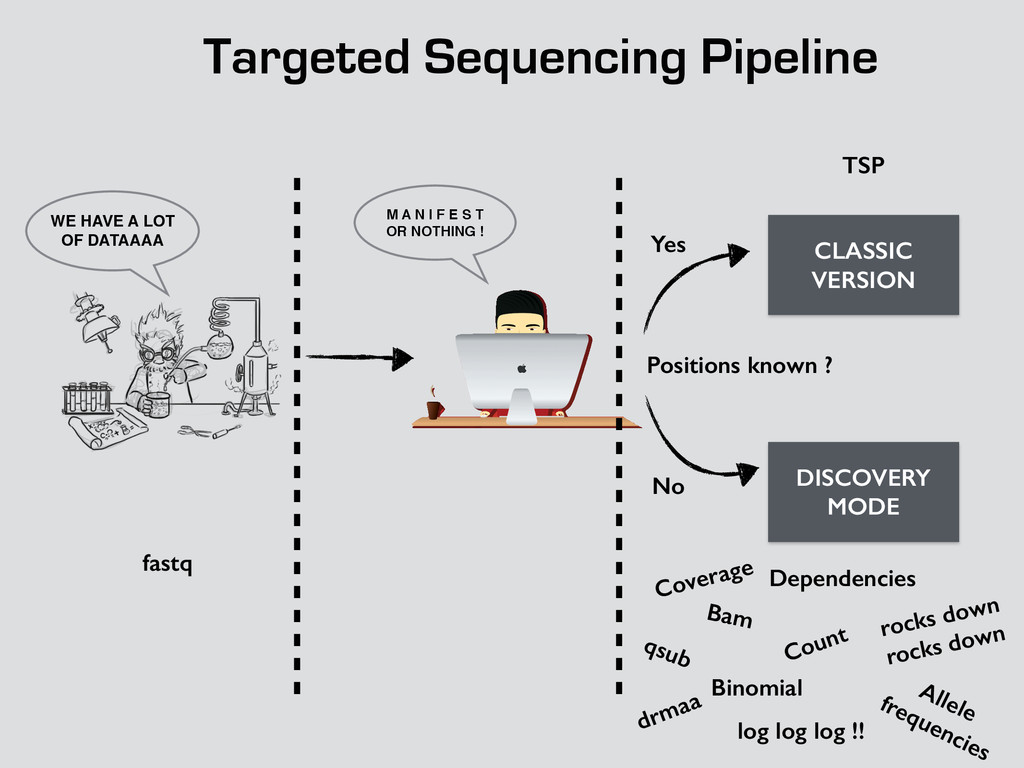
T! OR NOTHING ! WE HAVE A LOT OF DATAAAA CLASSIC VERSION DISCOVERY MODE Positions known ? Yes No fastq TSP Bam Count Binomial Dependencies Coverage Allele frequencies log log log !! qsub drmaa rocks down rocks down
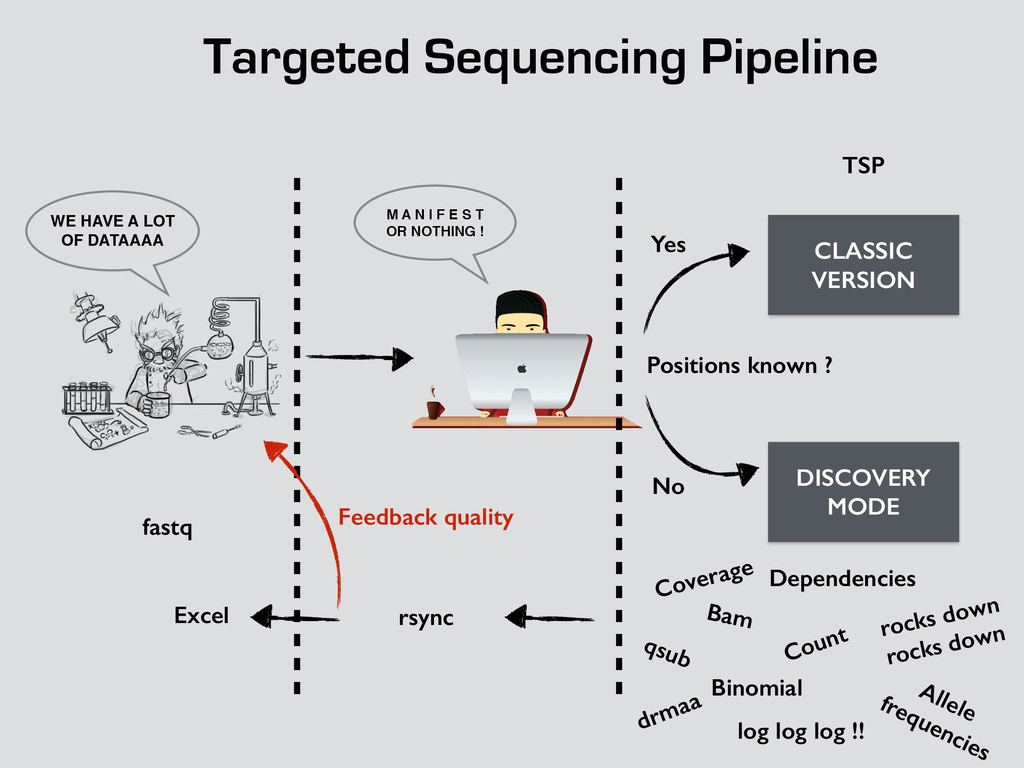
T! OR NOTHING ! WE HAVE A LOT OF DATAAAA CLASSIC VERSION DISCOVERY MODE Positions known ? Yes No fastq TSP Bam Count Binomial Dependencies Coverage Allele frequencies log log log !! qsub drmaa rocks down rocks down rsync Excel Feedback quality
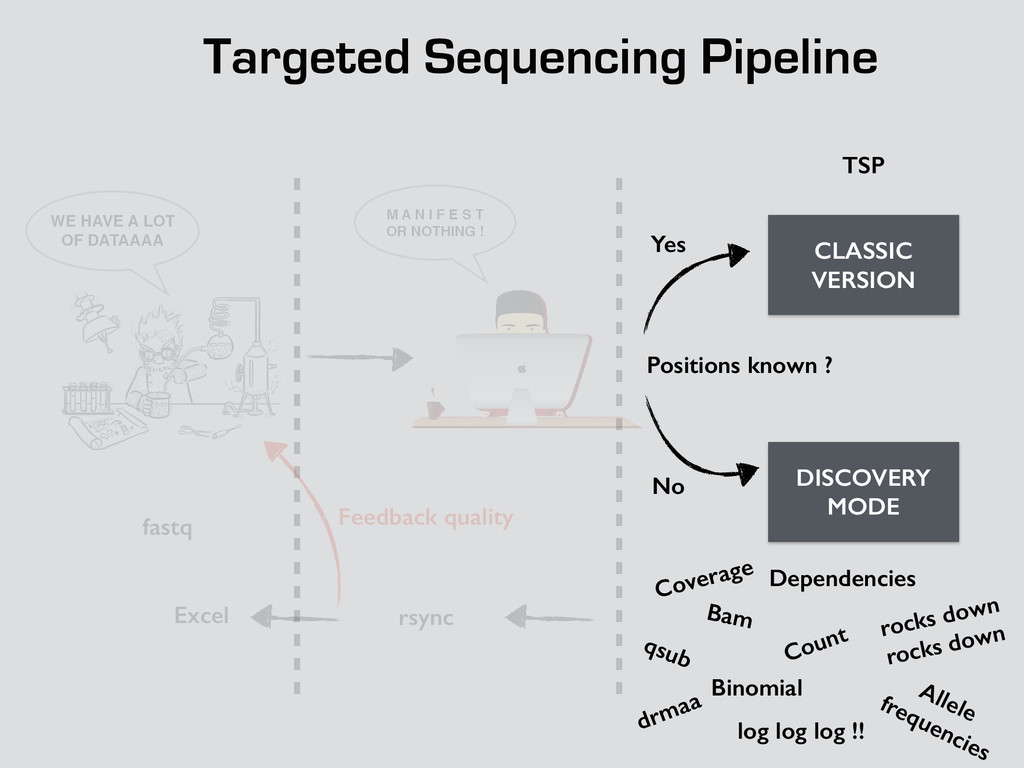
T! OR NOTHING ! WE HAVE A LOT OF DATAAAA CLASSIC VERSION DISCOVERY MODE Positions known ? Yes No fastq TSP Bam Count Binomial Dependencies Coverage Allele frequencies log log log !! qsub drmaa rocks down rocks down rsync Excel Feedback quality
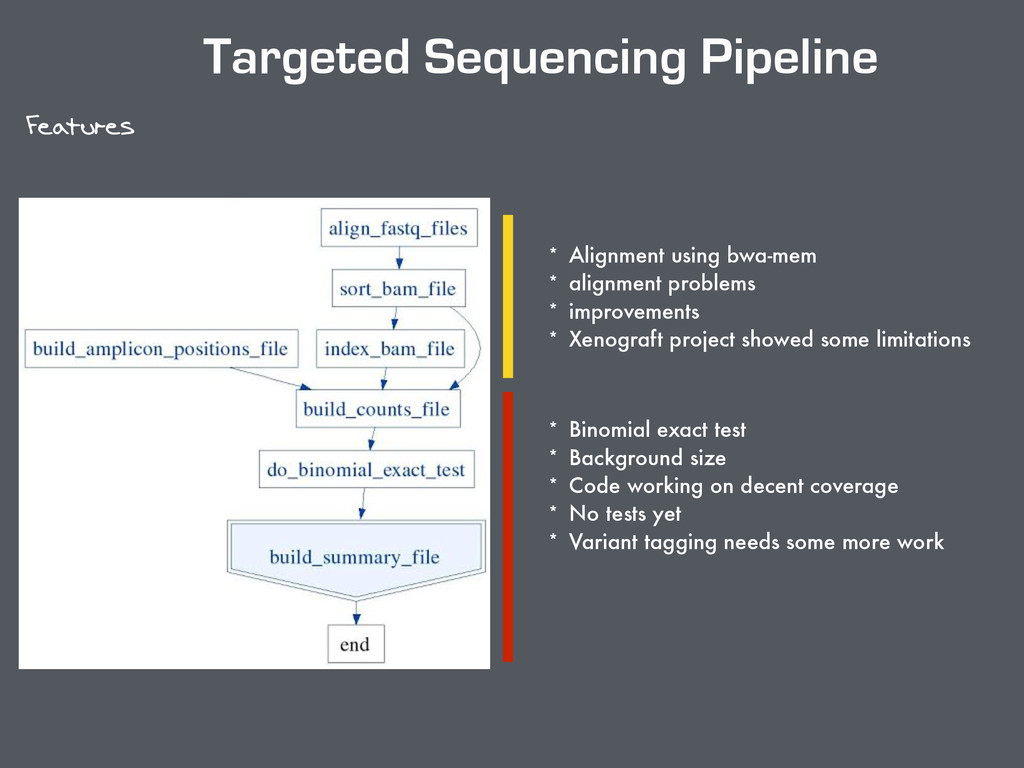
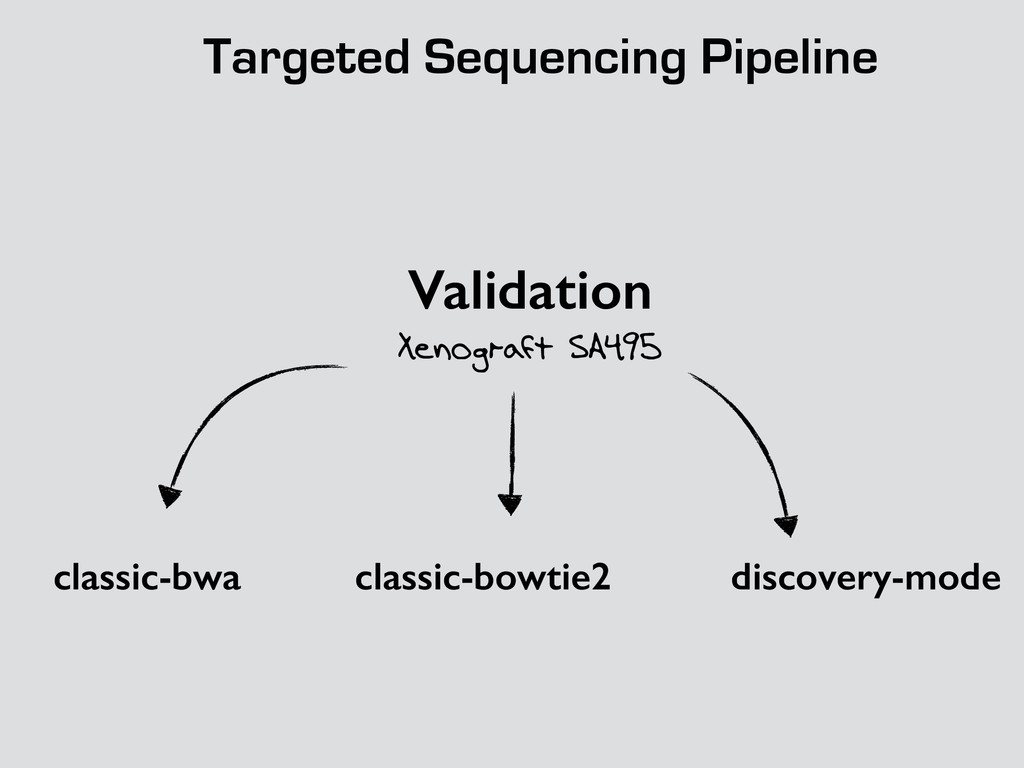
problems * improvements * Xenograft project showed some limitations * Binomial exact test * Background size * Code working on decent coverage * No tests yet * Variant tagging needs some more work
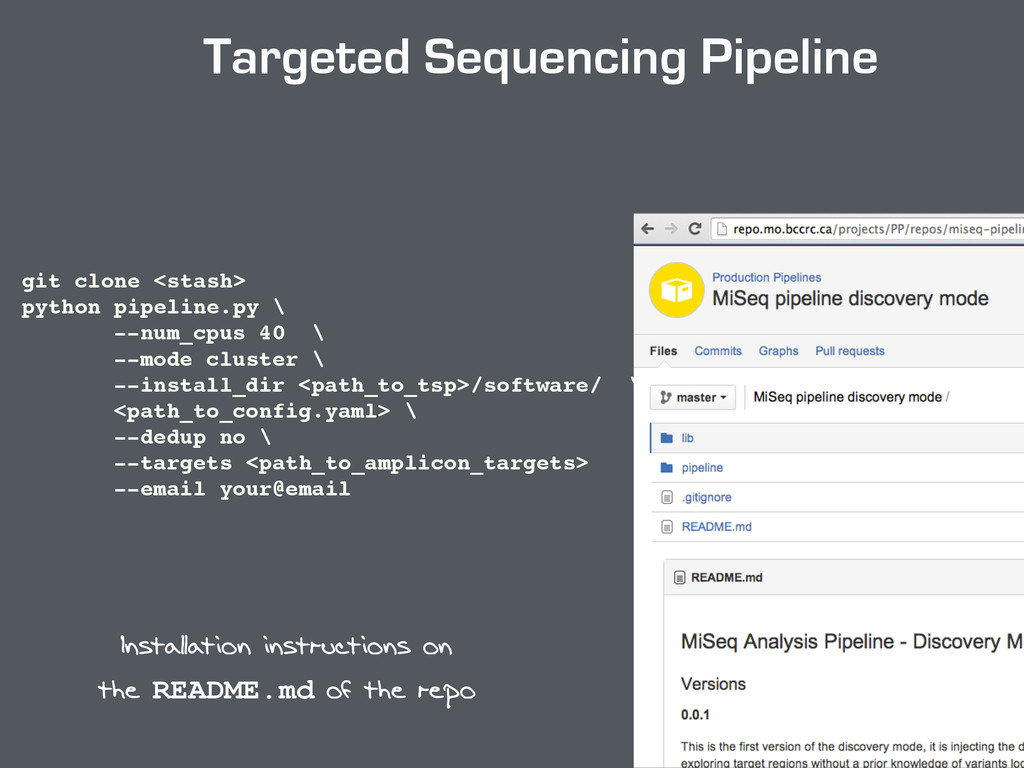
pipeline.py \ —num_cpus 10 \ —mode cluster \ —install_dir <path_to_miseq>/software \ <path_to_config_file> Limitations : ! - Manifest file - positions format - limited to point mutations ! ! If you want to run this version : ! - create_config_file_for_miseqpipeline.py One single code that generates all needed formats to run the pipeline
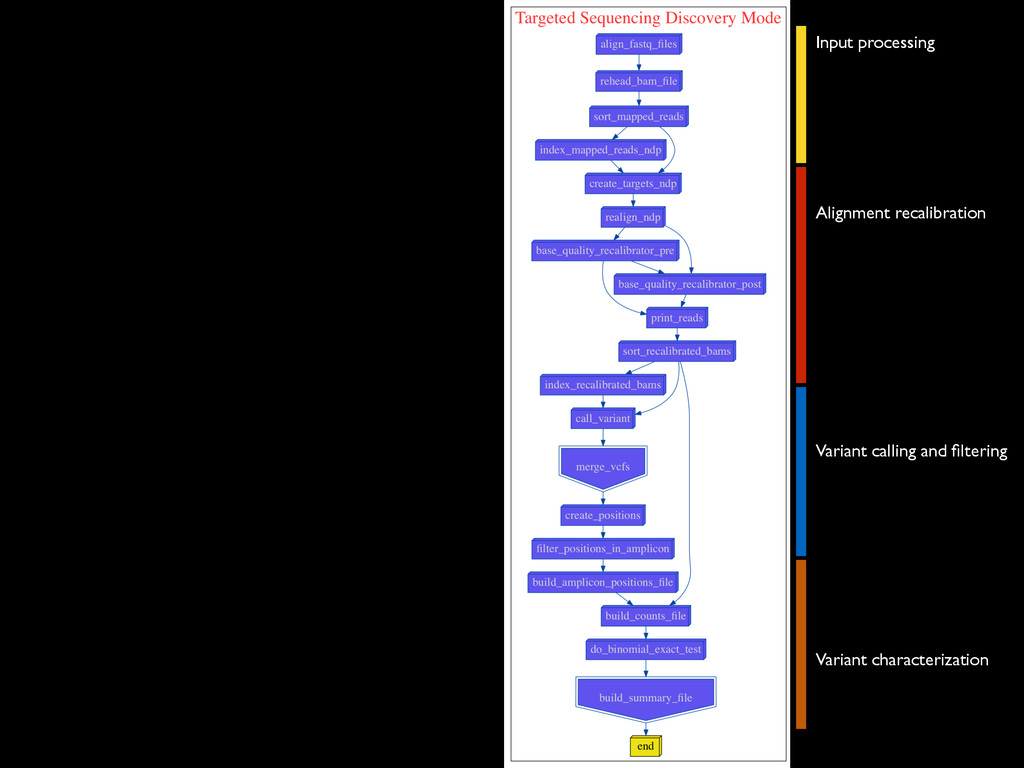
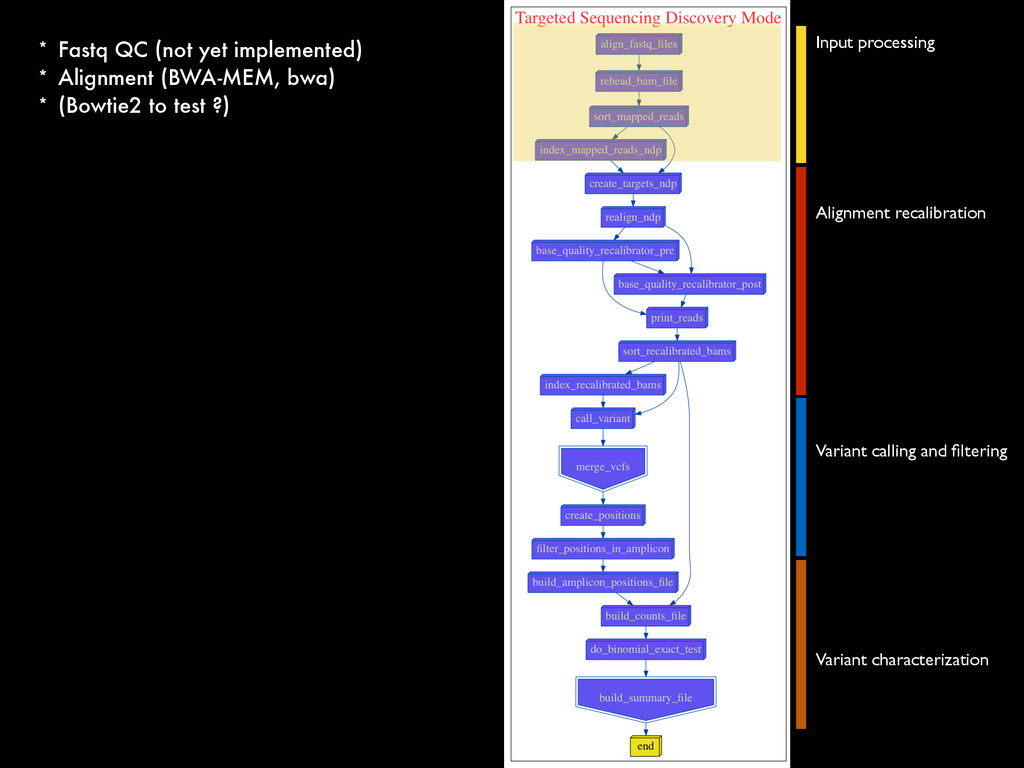
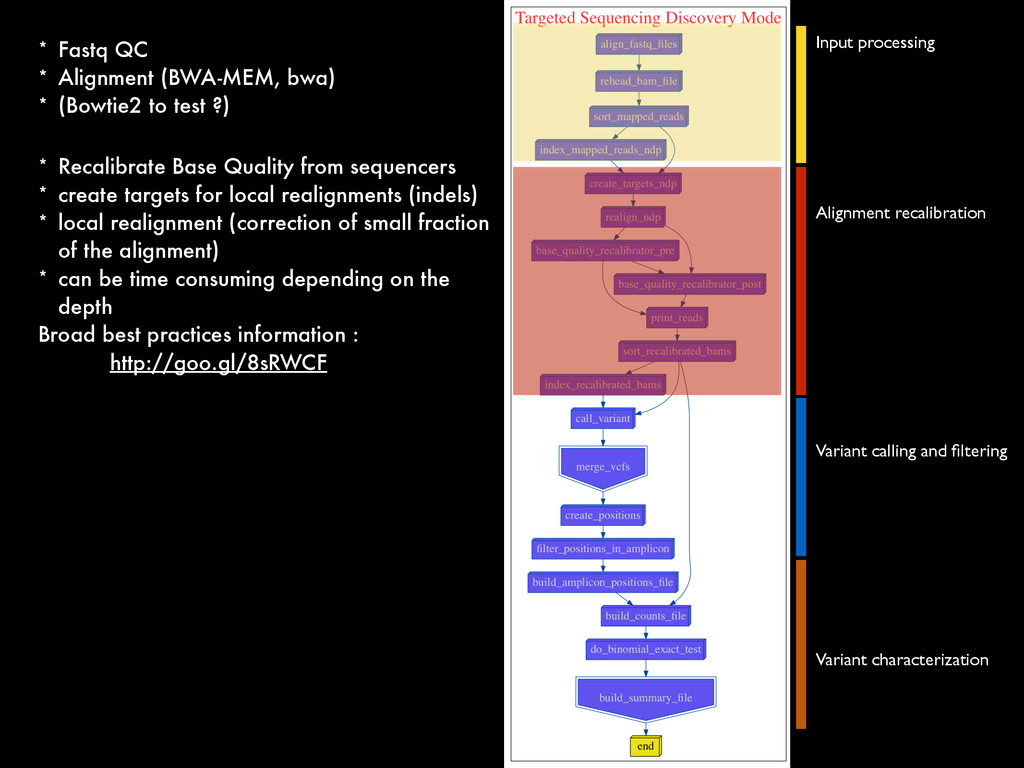
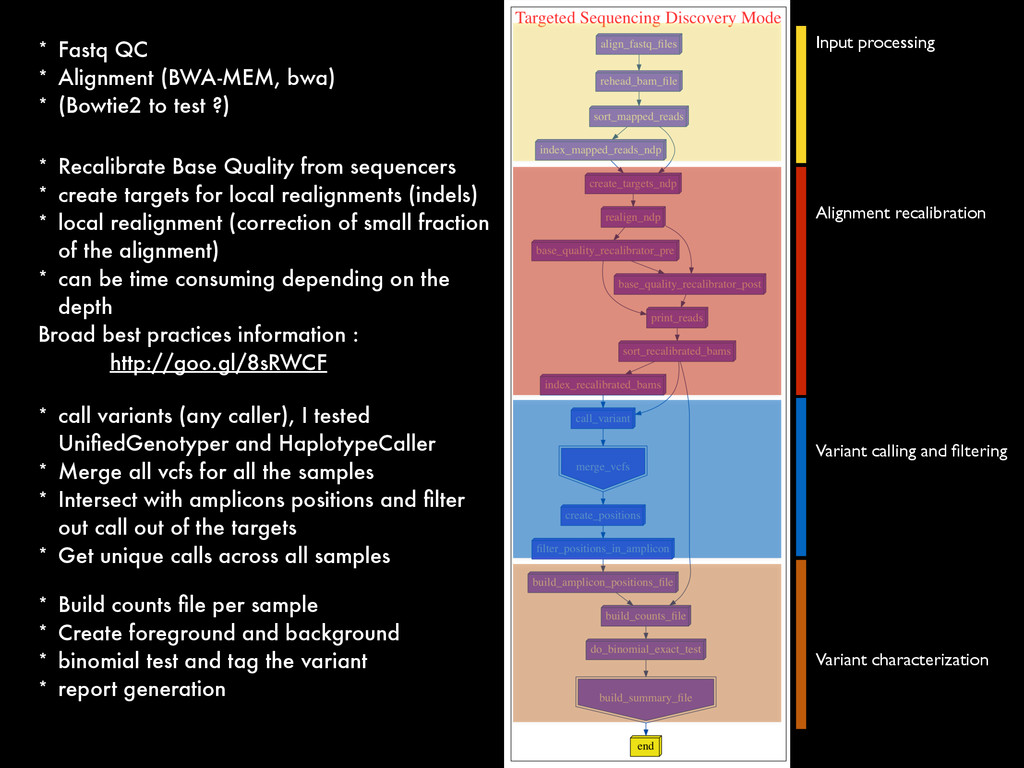
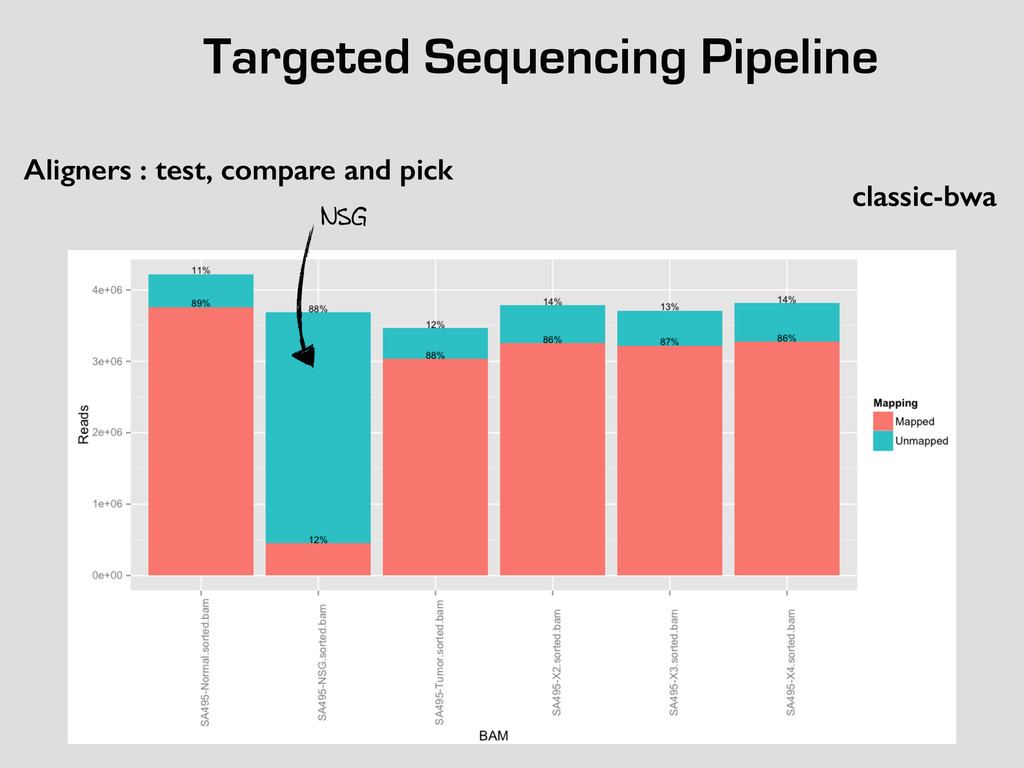
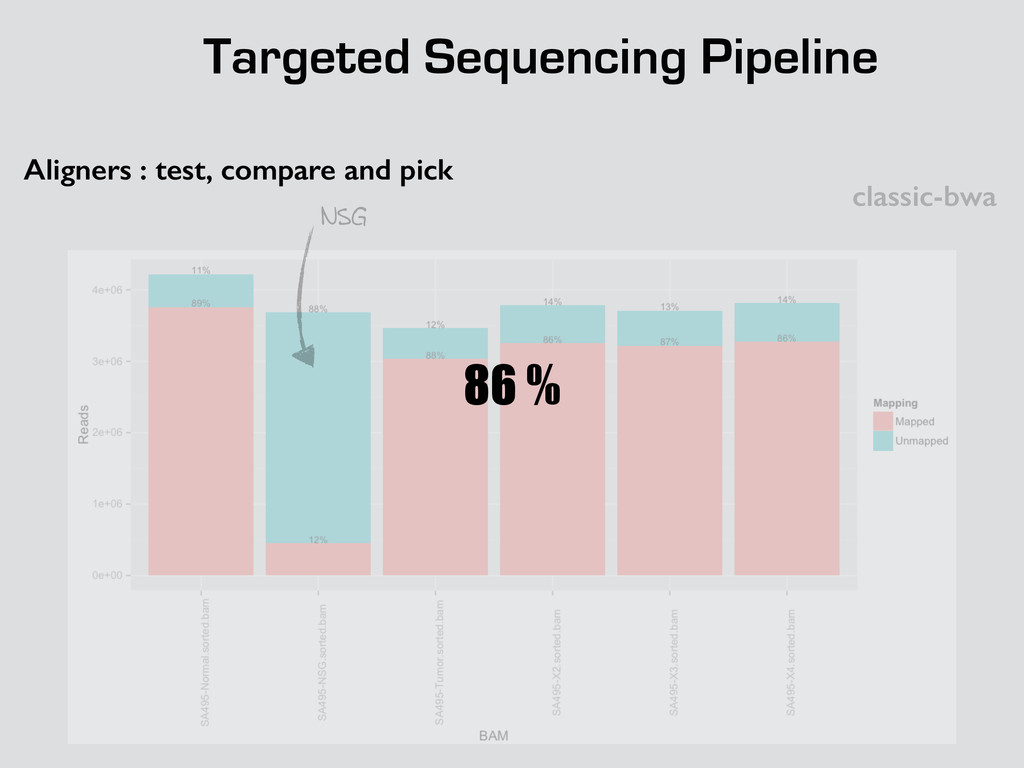
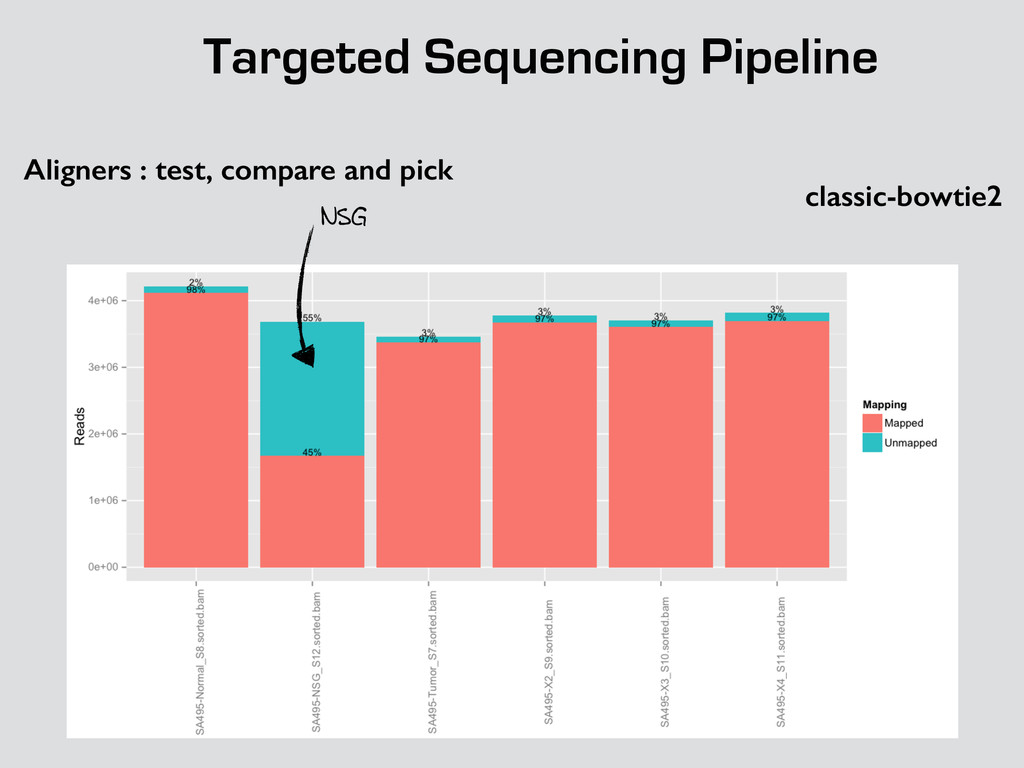
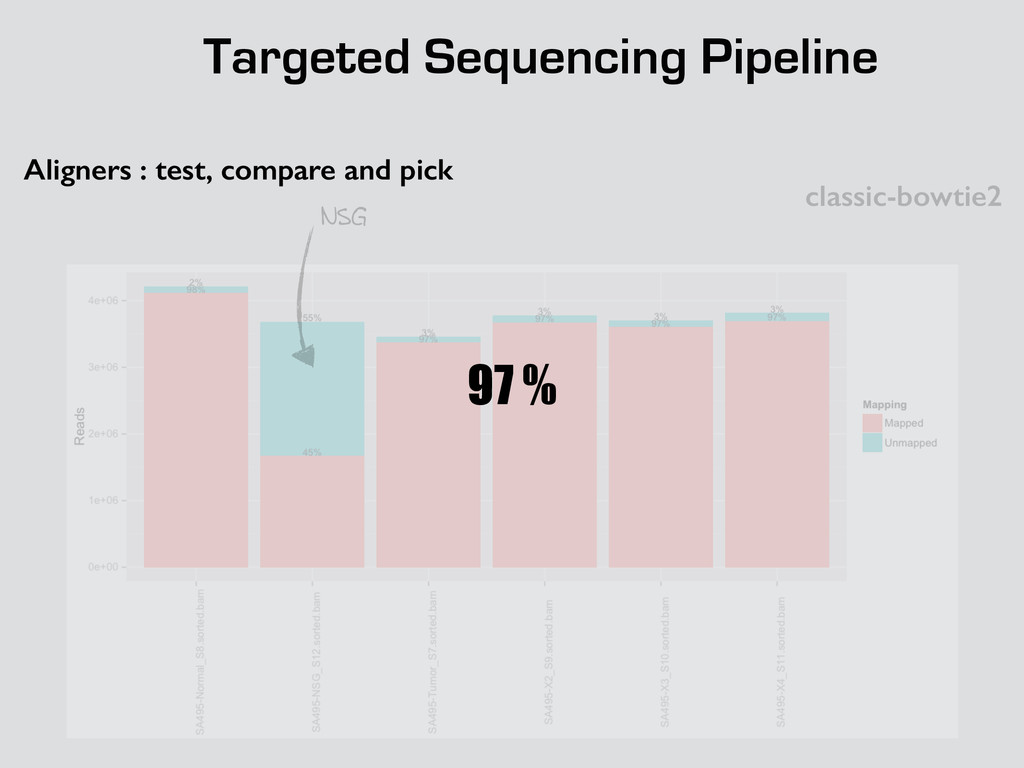
* Recalibrate Base Quality from sequencers * create targets for local realignments (indels) * local realignment (correction of small fraction of the alignment) * can be time consuming depending on the depth Broad best practices information : http://goo.gl/8sRWCF * Fastq QC * Alignment (BWA-MEM, bwa) * (Bowtie2 to test ?)
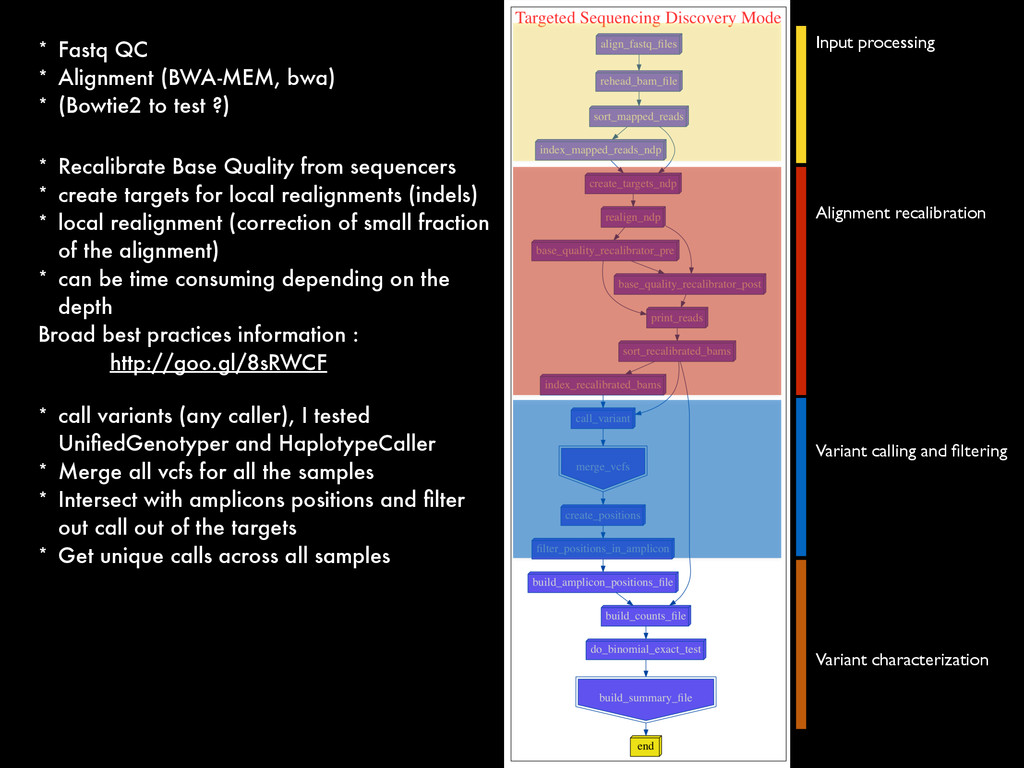
* Recalibrate Base Quality from sequencers * create targets for local realignments (indels) * local realignment (correction of small fraction of the alignment) * can be time consuming depending on the depth Broad best practices information : http://goo.gl/8sRWCF * Fastq QC * Alignment (BWA-MEM, bwa) * (Bowtie2 to test ?) * call variants (any caller), I tested UnifiedGenotyper and HaplotypeCaller * Merge all vcfs for all the samples * Intersect with amplicons positions and filter out call out of the targets * Get unique calls across all samples
* Recalibrate Base Quality from sequencers * create targets for local realignments (indels) * local realignment (correction of small fraction of the alignment) * can be time consuming depending on the depth Broad best practices information : http://goo.gl/8sRWCF * Fastq QC * Alignment (BWA-MEM, bwa) * (Bowtie2 to test ?) * call variants (any caller), I tested UnifiedGenotyper and HaplotypeCaller * Merge all vcfs for all the samples * Intersect with amplicons positions and filter out call out of the targets * Get unique calls across all samples * Build counts file per sample * Create foreground and background * binomial test and tag the variant * report generation
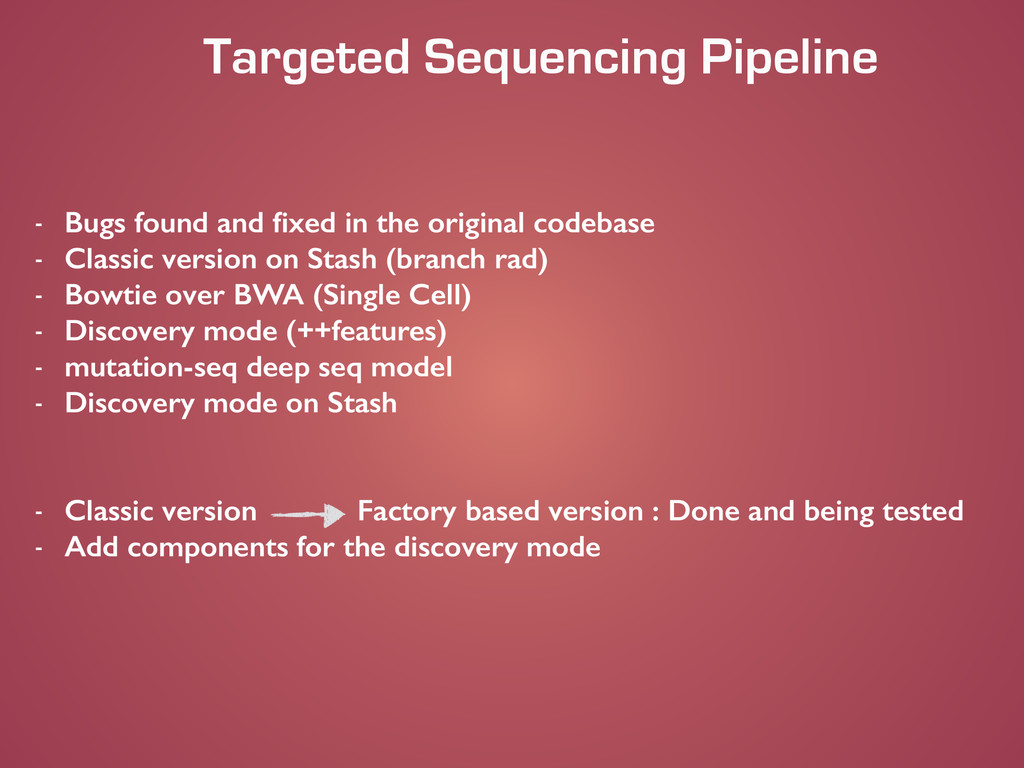
original codebase - Classic version on Stash (branch rad) - Bowtie over BWA (Single Cell) - Discovery mode (++features) - mutation-seq deep seq model - Discovery mode on Stash ! ! - Classic version Factory based version : Done and being tested - Add components for the discovery mode
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Targeted Sequencing Pipeline git clone http://[email protected]/scm/pp/miseq-pipeline.git cd miseq-pipeline/pipeline/code ! python](https://files.speakerdeck.com/presentations/f4660b4000f401329bf92ec7aa488f39/slide_12.jpg){kind=link}
![Targeted Sequencing Pipeline git clone http://[email protected]/scm/pp/miseq-pipeline.git cd miseq-pipeline/pipeline/code ! python](https://files.speakerdeck.com/presentations/f4660b4000f401329bf92ec7aa488f39/slide_13.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
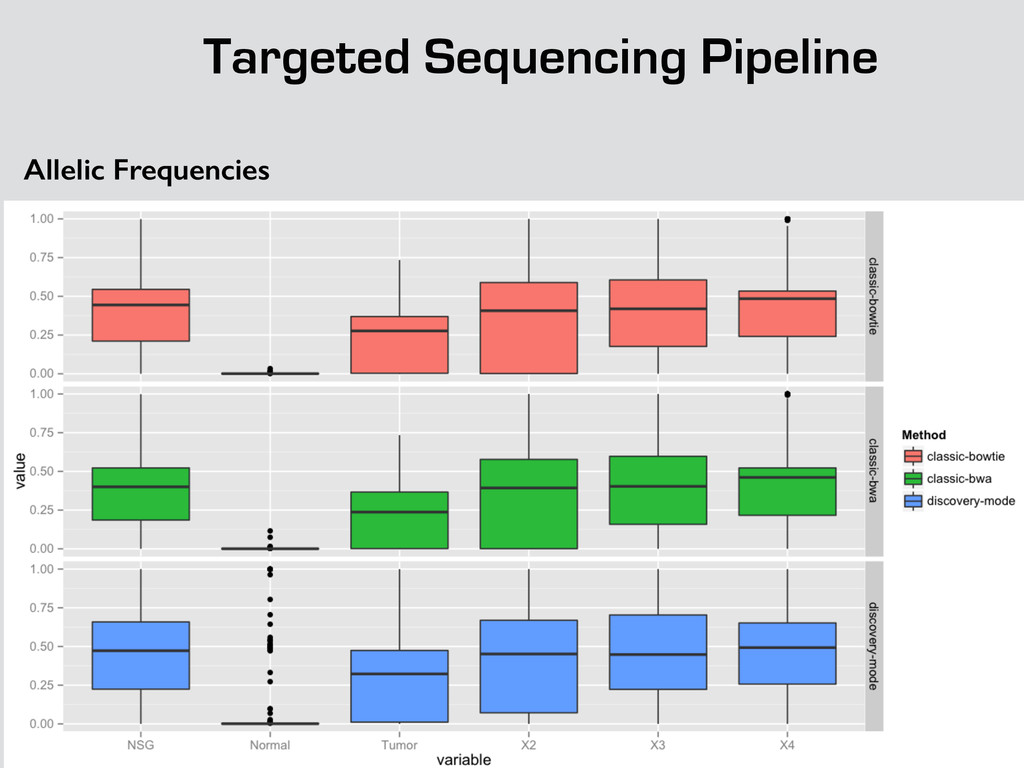{kind=link}
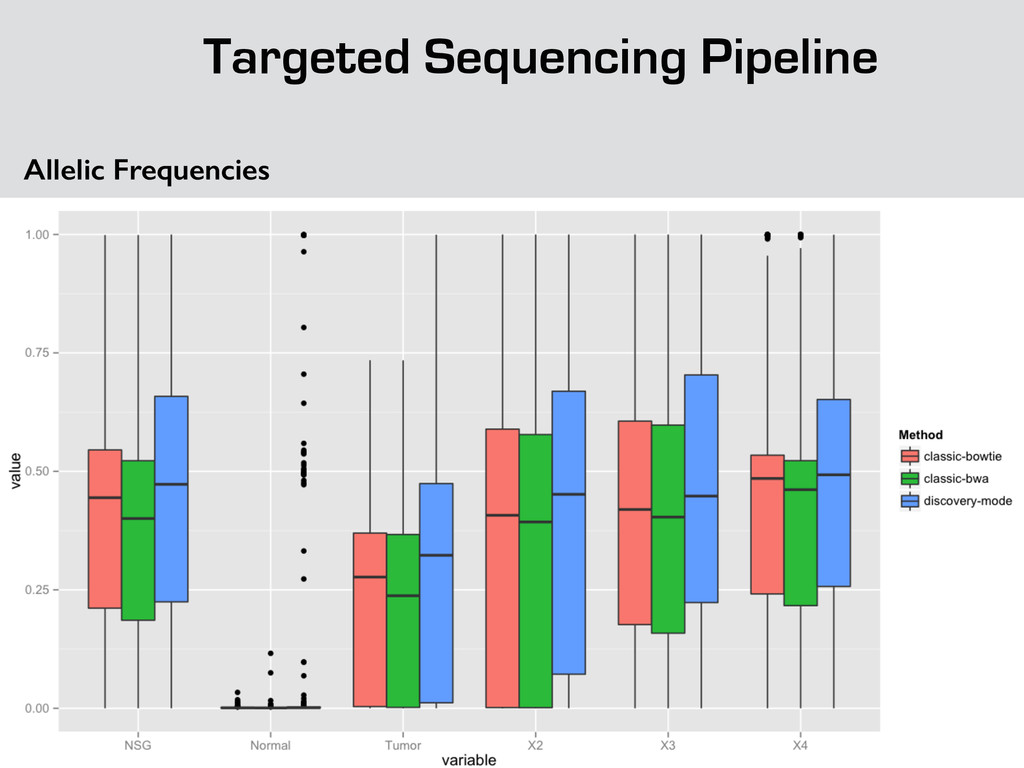{kind=link}
{kind=link}
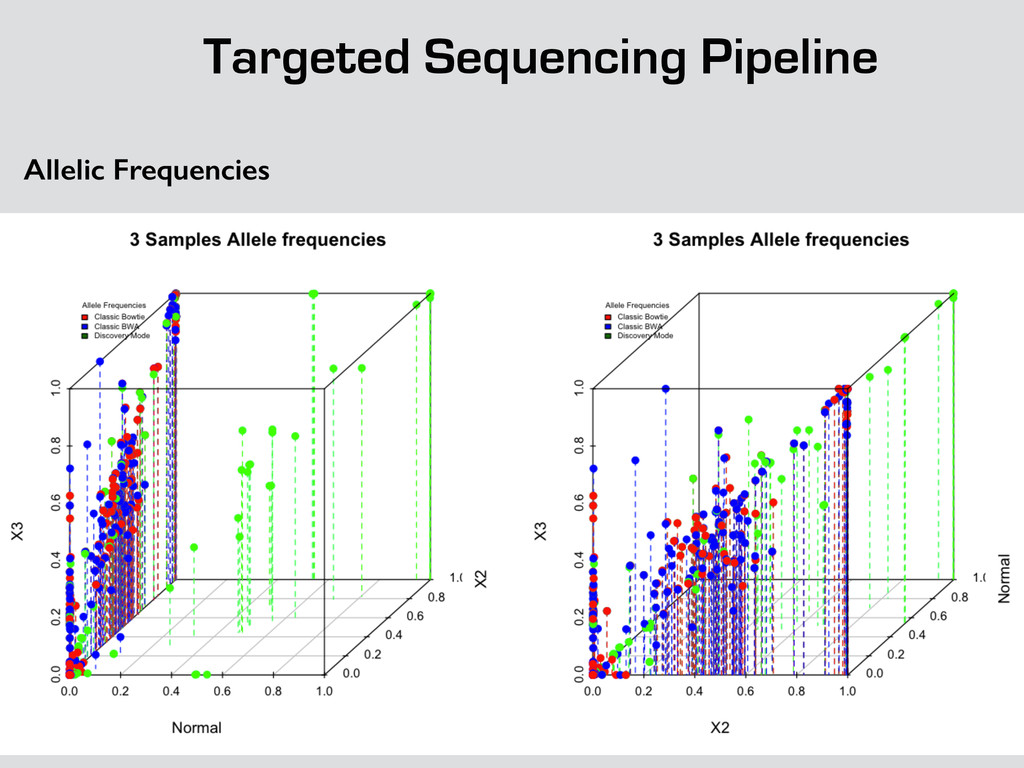{kind=link}
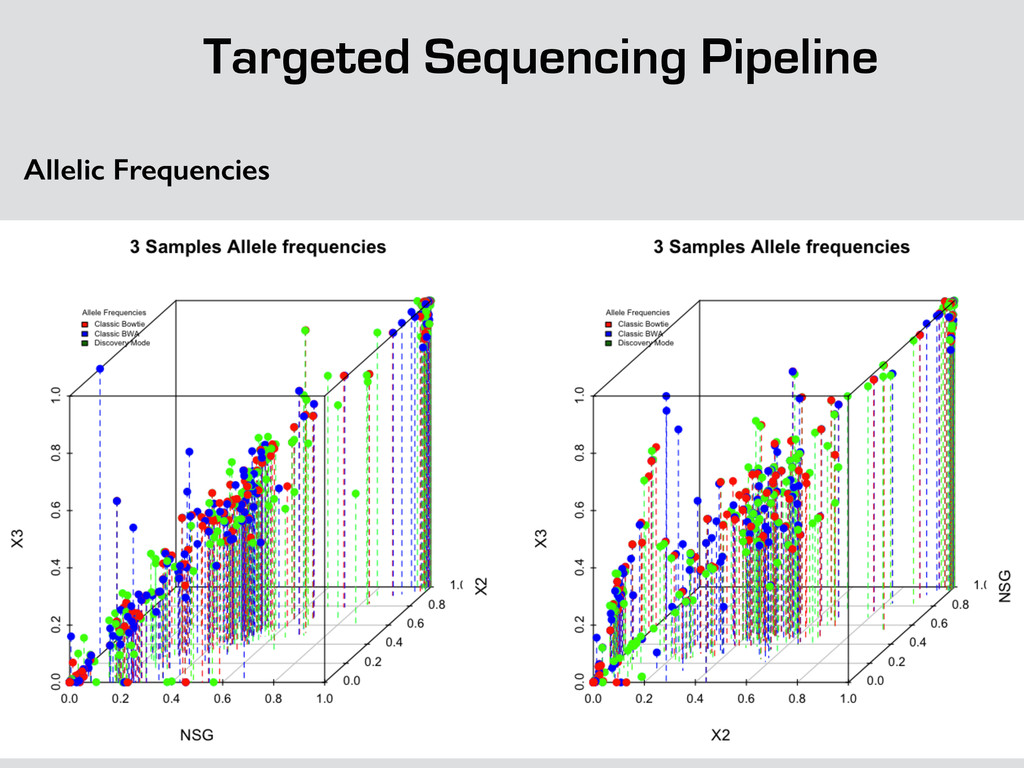{kind=link}
{kind=link}
{kind=link}