• Your mileage will vary depending on repeat type • Centromeres still a challenge • Evidence of systematic error/coverage gaps • Human-level heterozygosity resolved • No polishing needed, consensus >Q50 • Data collection is the bottleneck • 30 hr/cell, 5 days for a 3g mammal • CCS consensus, ≈6k cpu hrs, ≈ 2 days on 128 cores • Canu asm, ≈2k cpu hrs, ≈ 0.5 day on 128 cores (<8 hrs on a cluster) Conclusions
• Adam Phillippy • Evan E. Eichler • Mitchell Vollger • Glennis Logsdon • Robert Grothe • Jonas Korlach • Zev Kronenberg • Paul Peluso • David Rank • Kevin Fengler • Sung Bong Shin • Tim Smith Acknowledgements
{kind=link}
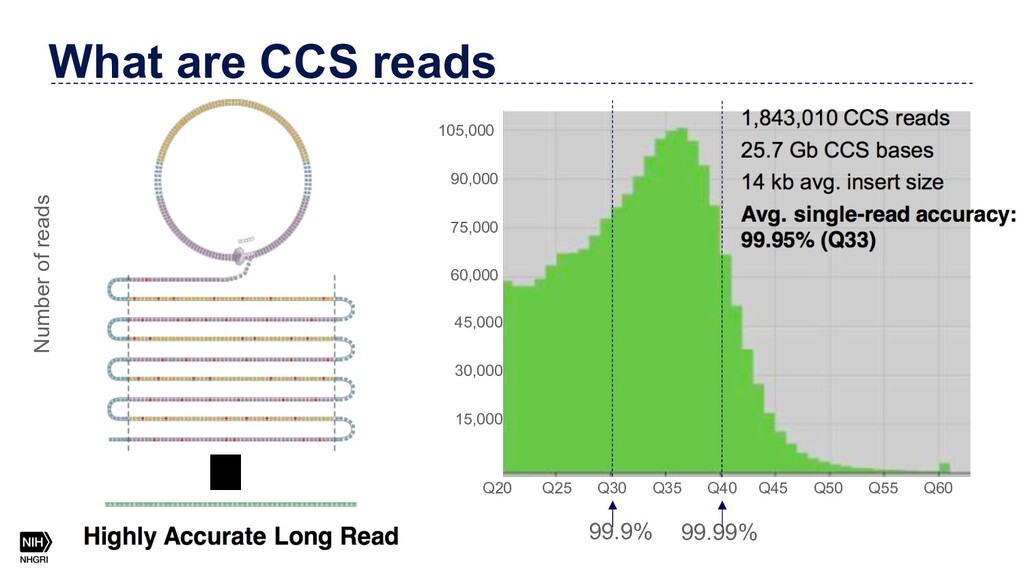{kind=link}
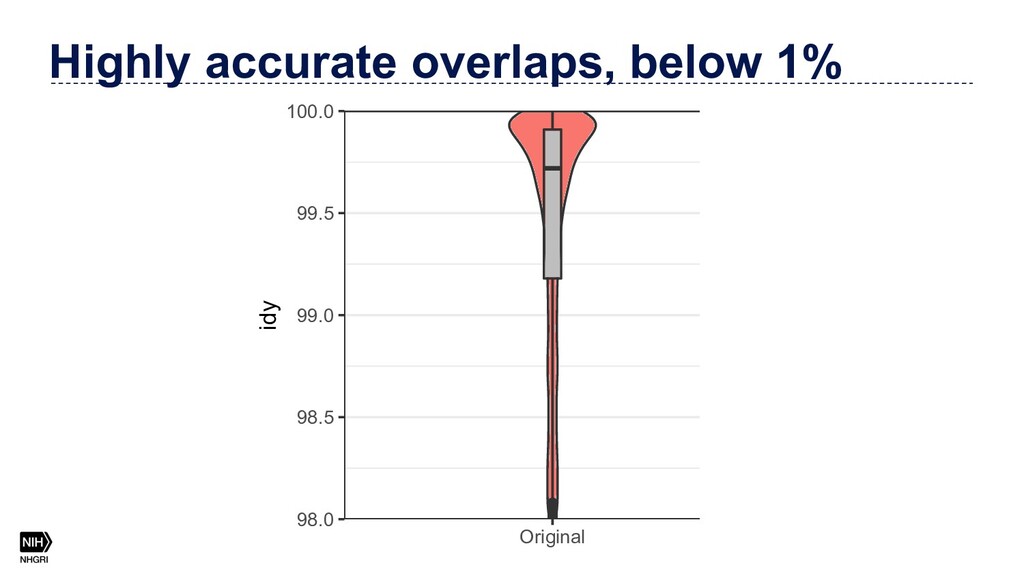{kind=link}
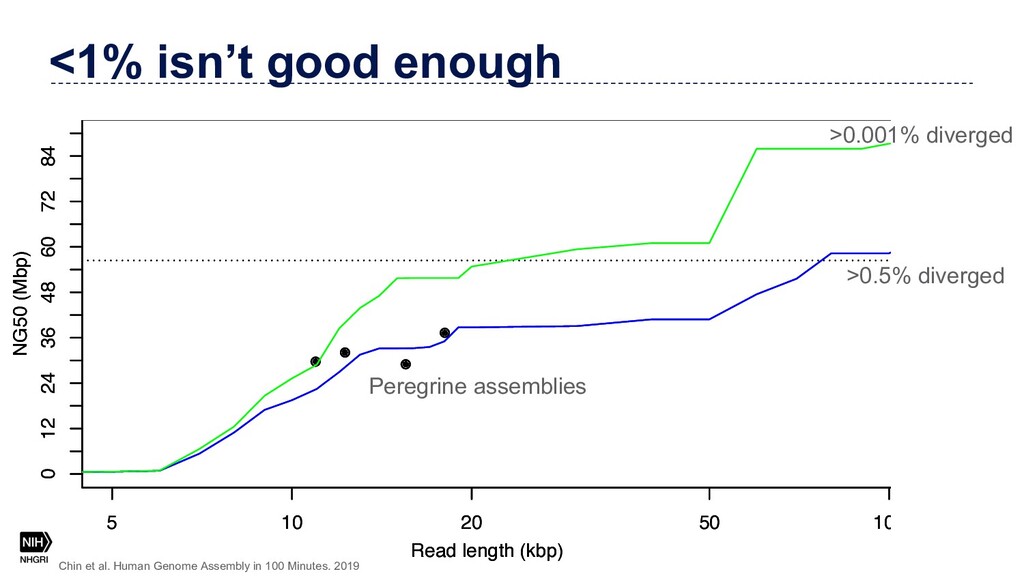{kind=link}
{kind=link}
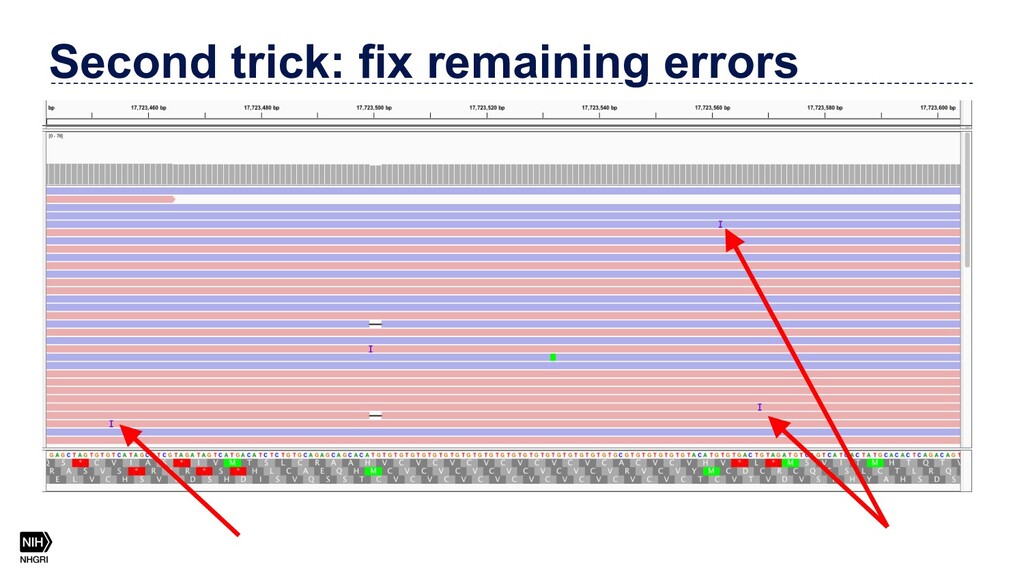{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}