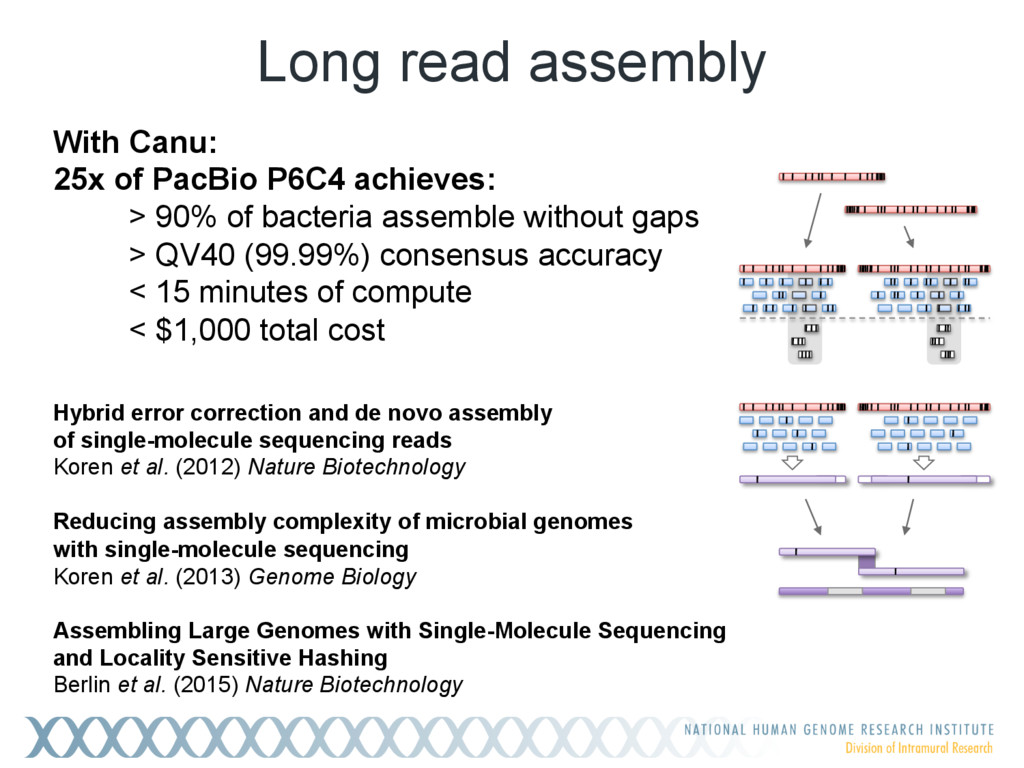
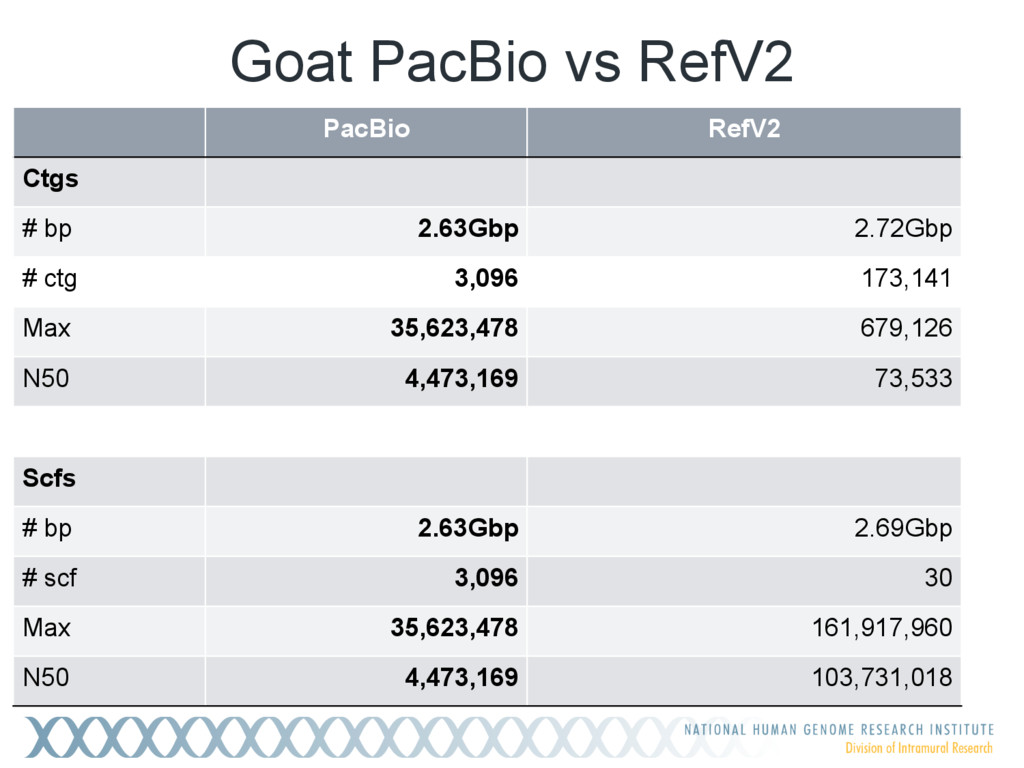
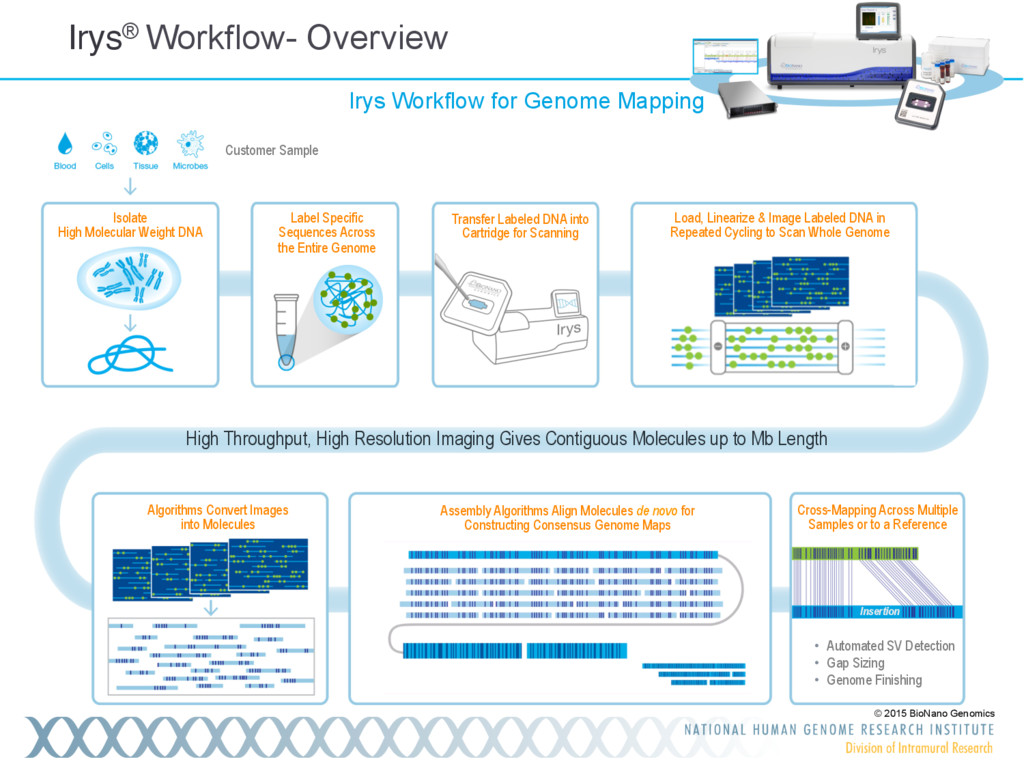
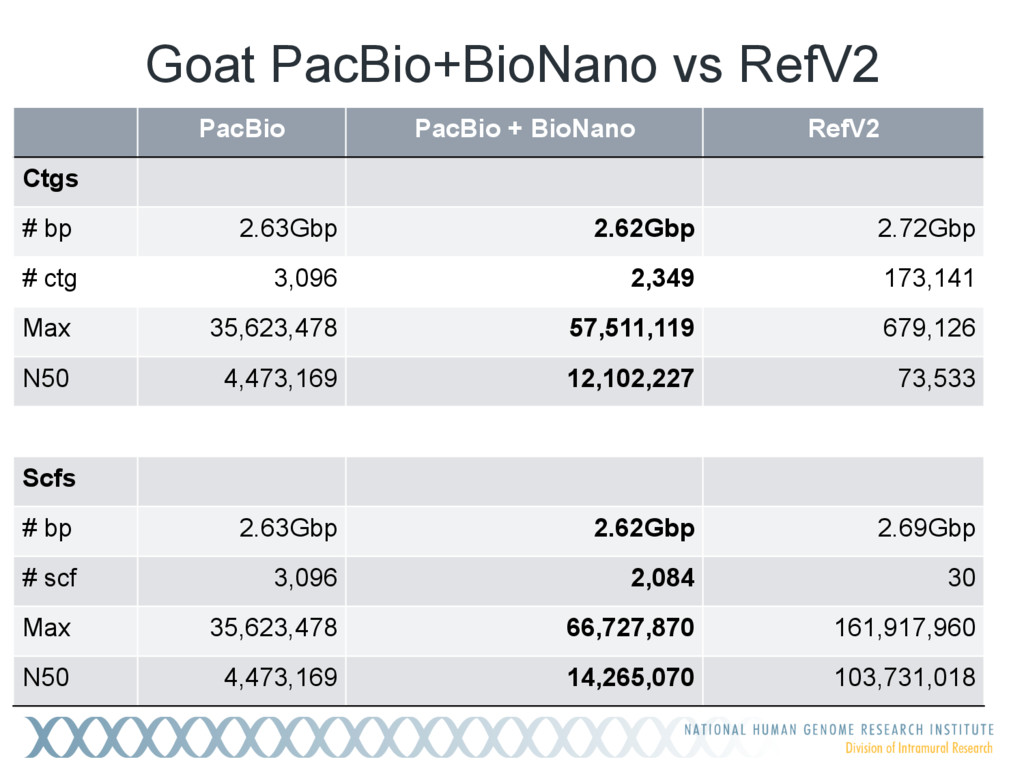
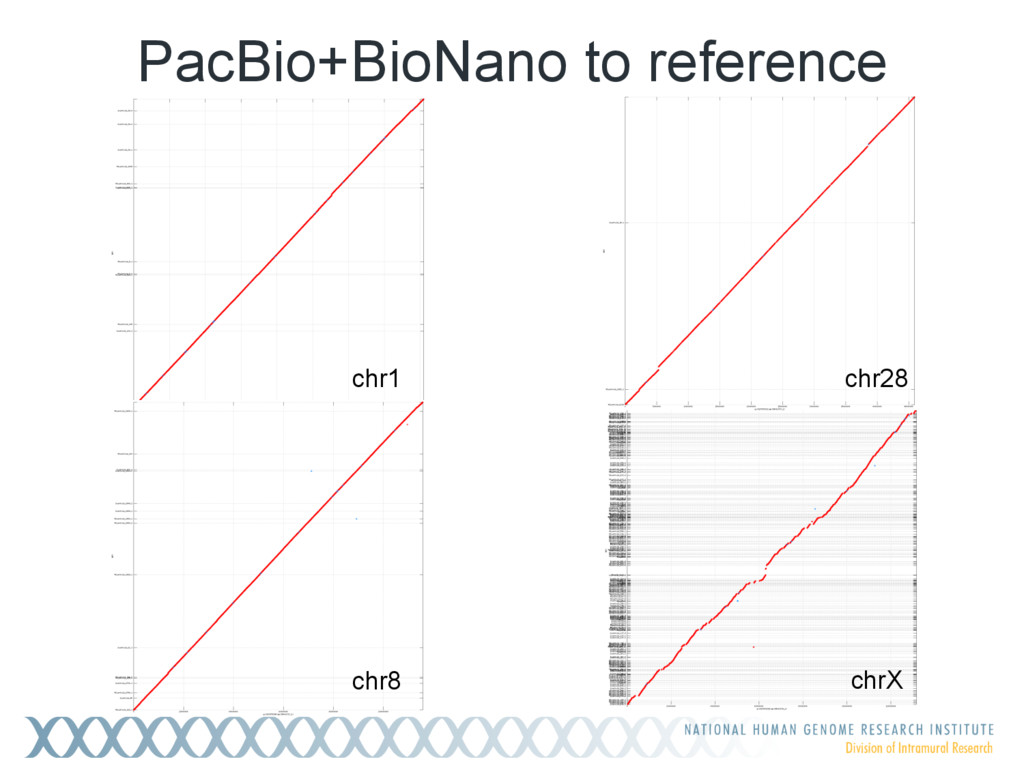
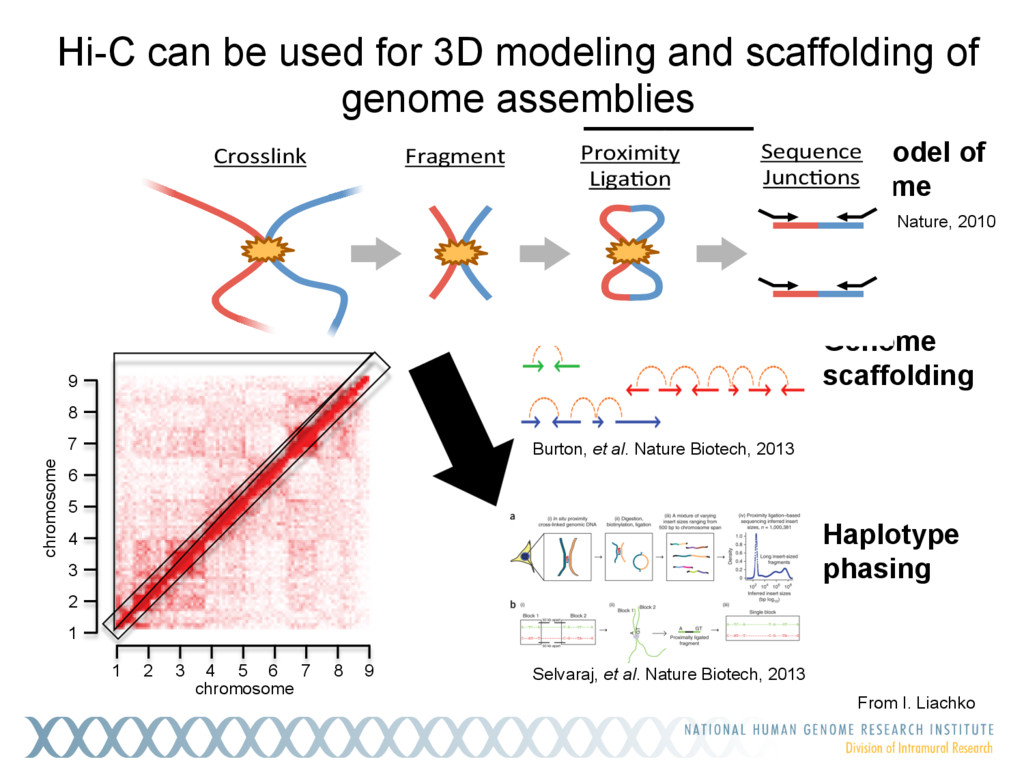
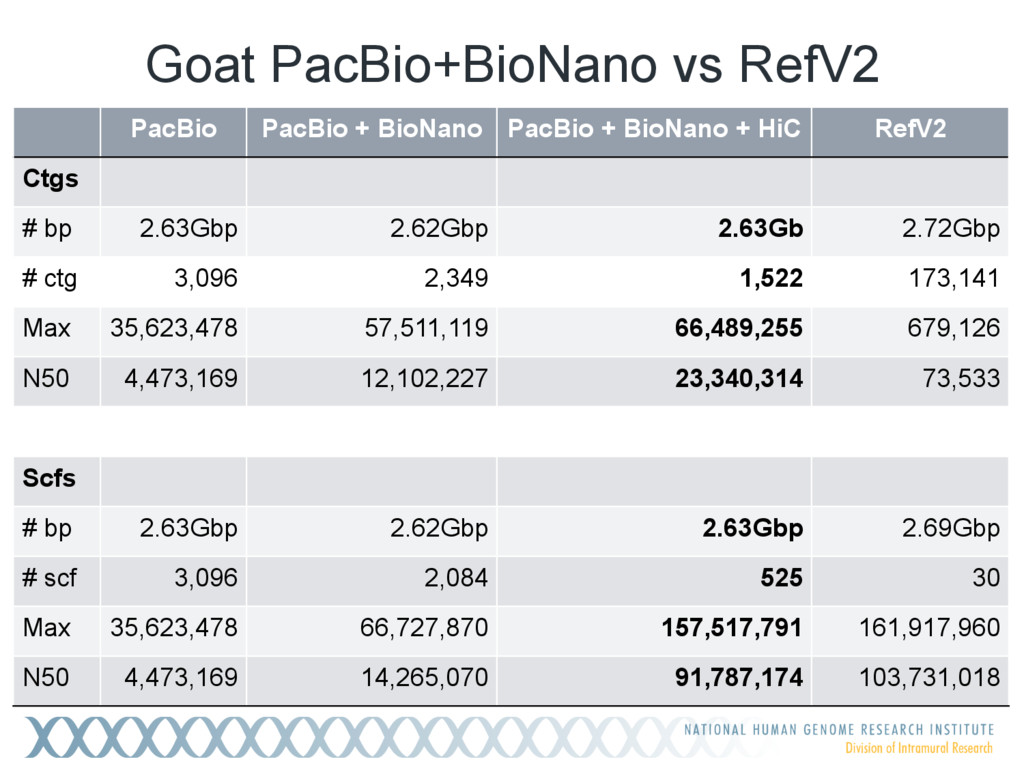
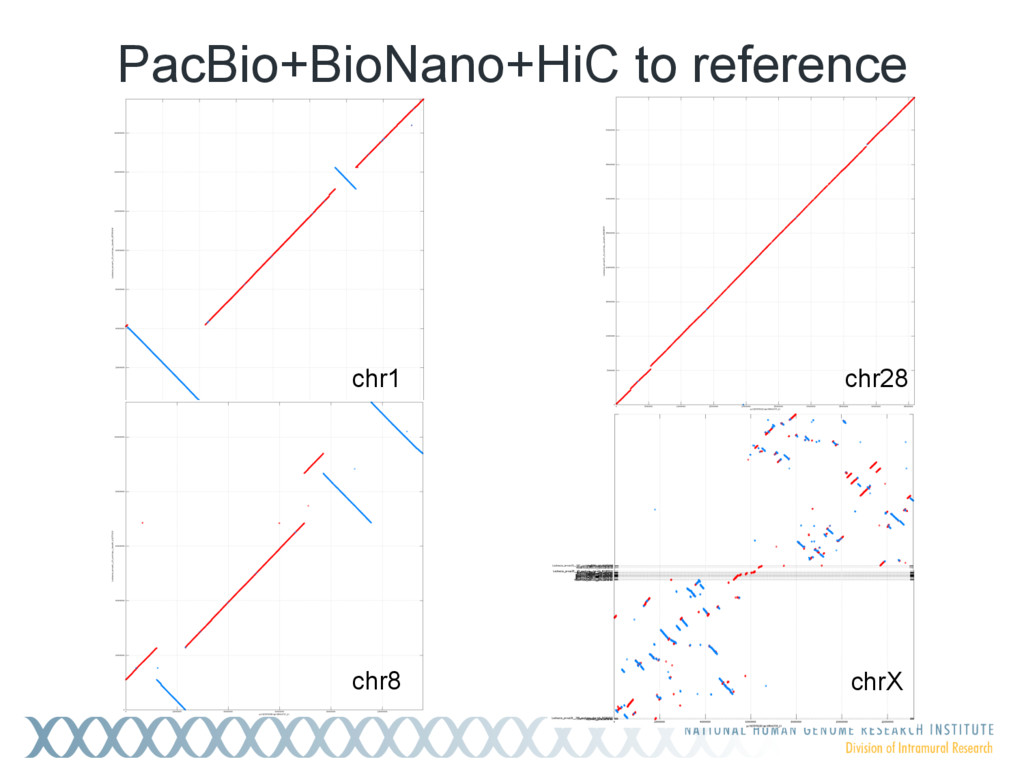
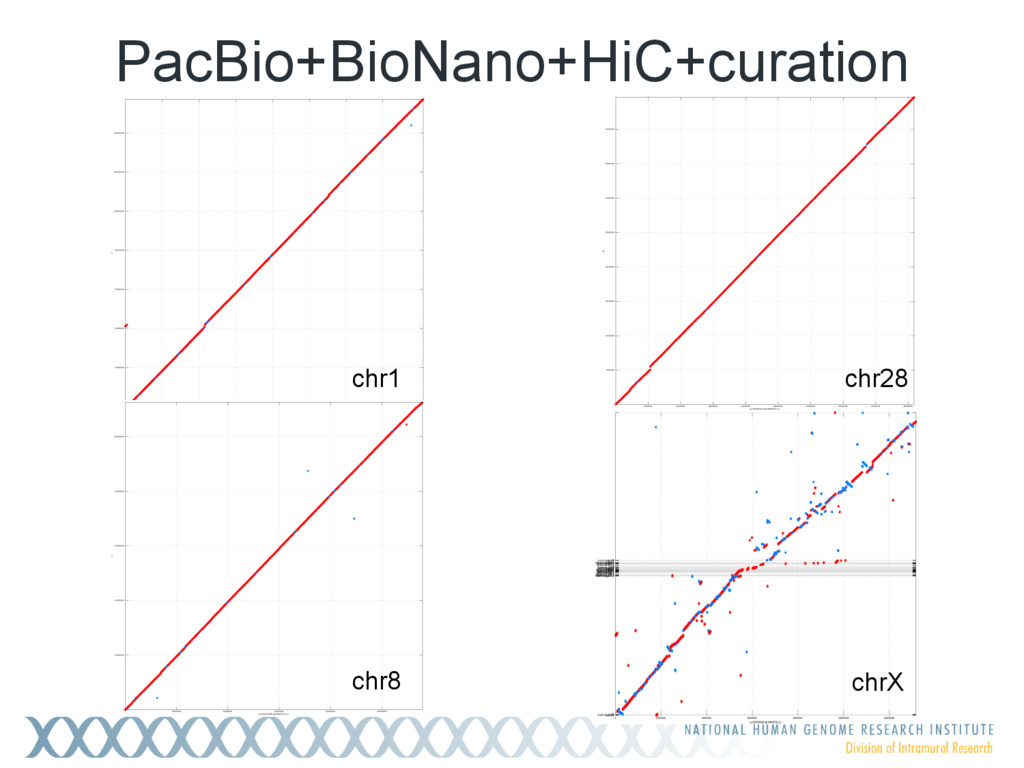
Single-molecule sequencing is now routinely used to assemble complete, high-quality microbial genomes, but these assembly methods have not scaled well to large genomes. To address this problem, we previously introduced the MinHash Alignment Process (MHAP) for overlapping single-molecule reads using probabilistic, locality-sensitive hashing. Integrating MHAP with Celera Assembler (CA) has enabled reference-grade assemblies of model organisms, revealing novel heterochromatic sequences and filling low-complexity gap sequences in the GRCh38 human reference genome. We have applied our methods to assemble the San Clemente goat genome. Combining single-molecule sequencing from Pacific Biosciences and BioNano Genomics generates and assembly that is over 150-fold more contiguous than the latest Capra hircus reference. In combination with Hi-C sequencing, the assembly surpasses reference assemblies, de novo, with minimal manual intervention. The autosomes are each assembled into a single scaffold. Our assembly provides a more complete gene reconstruction, better alignments with Goat 52k chip, and improved allosome reconstruction. In addition to providing increased continuity of sequence, our assembly achieves a higher BUSCO completion score (84%) than the existing goat reference assembly suggesting better quality annotation of gene models. Our results demonstrate that single-molecule sequencing can produce near-complete eukaryotic genomes at modest cost and minimal manual effort.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}