PAGXXVI combined presentations from Genome Informatics section outlining the trio binning strategy to produce complete haplotypes from a single genome.
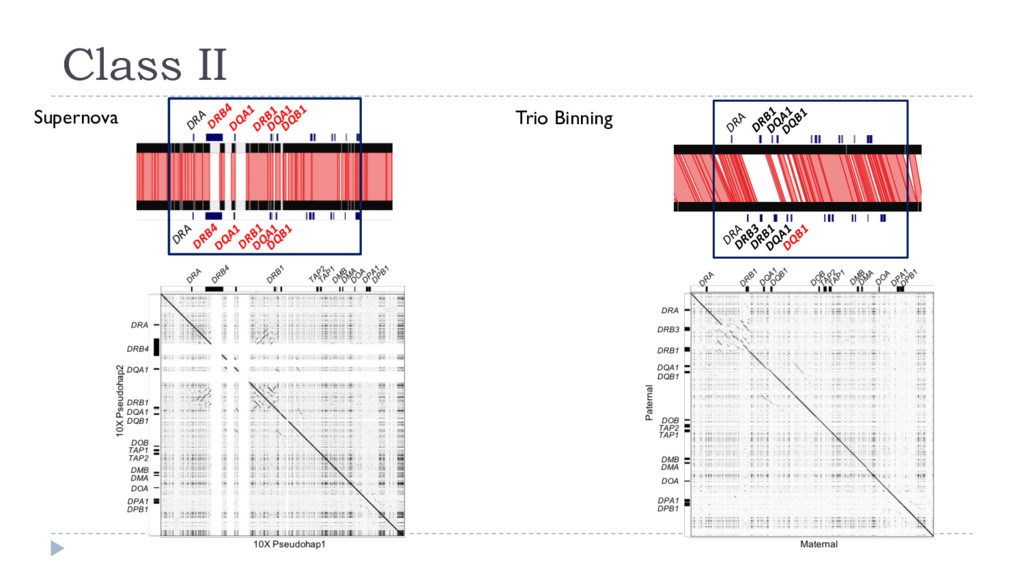
Heterozygosity important for fitness } Mixture of homozygous and collapsed heterozygous regions } Incomplete phasing } No association of blocks to a haplotype } Short phase blocks } Missed diverged heterozygous regions
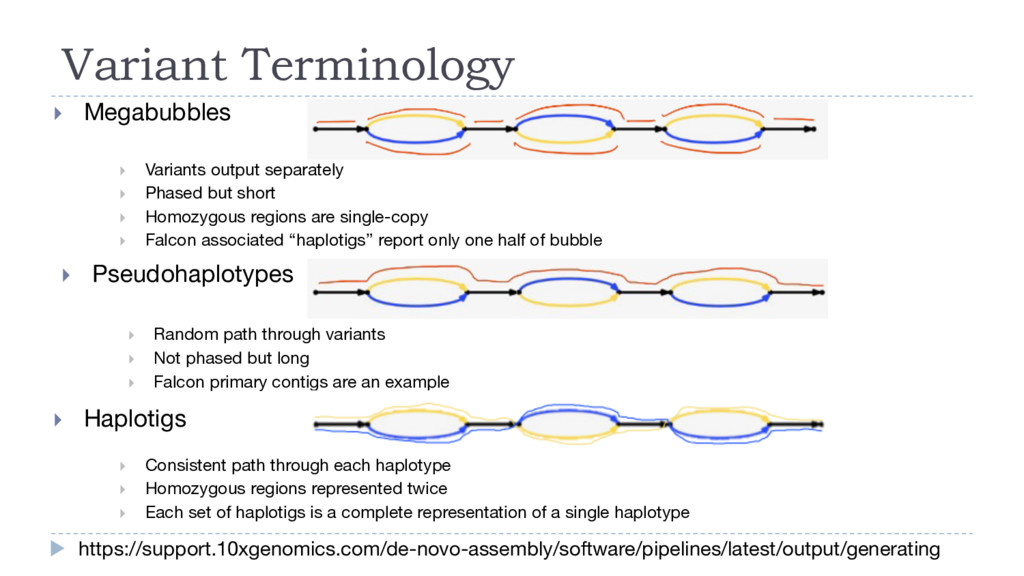
} Homozygous regions are single-copy } Falcon associated “haplotigs” report only one half of bubble Variant Terminology https://support.10xgenomics.com/de-novo-assembly/software/pipelines/latest/output/generating } Pseudohaplotypes } Random path through variants } Not phased but long } Falcon primary contigs are an example } Haplotigs } Consistent path through each haplotype } Homozygous regions represented twice } Each set of haplotigs is a complete representation of a single haplotype
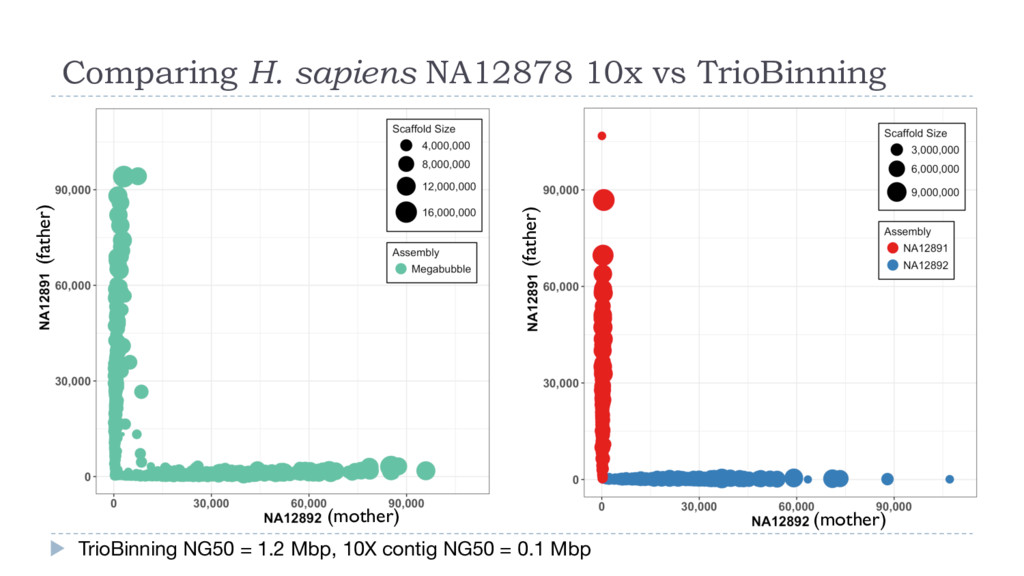
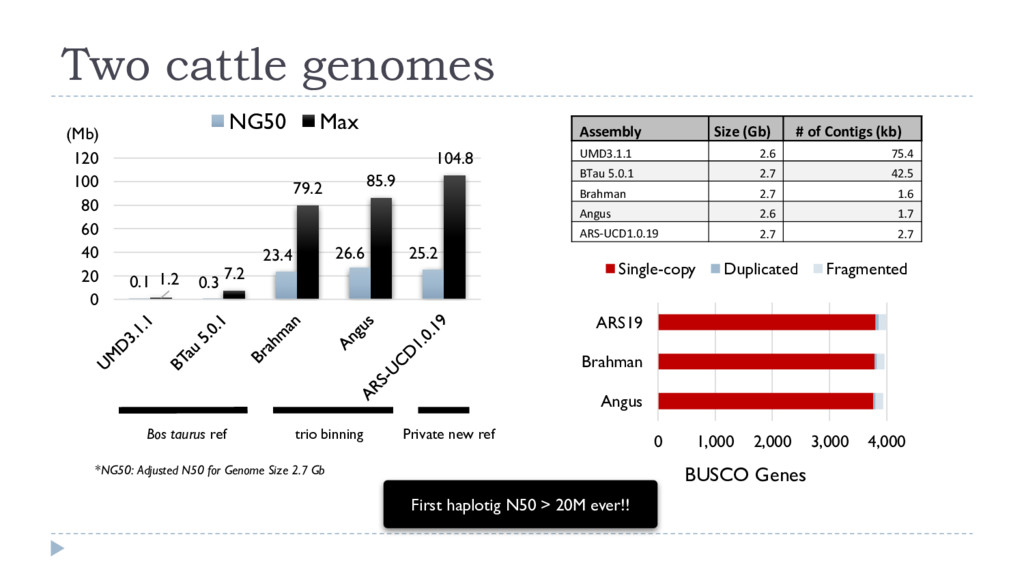
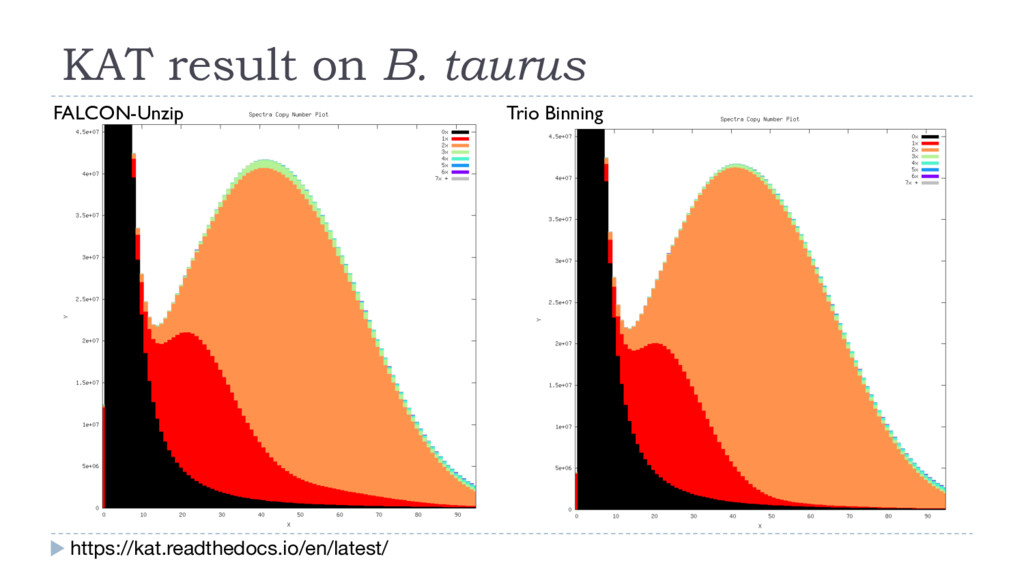
density Read length Trio Binning Dam (Brahman) haplotigs Sire (Angus) haplotigs 49.6% (67.3x) 10.9 kb 49.3% (66.9x) 11.7 kb 1.1% (1.4x), avg 1.3 kb canu • K-mer profiling of each parent (Illumina, 60x) Dam k-mers Sire k-mers • K-mer profiling of the F1 (PacBio, 120x) Angus x Brahman F1 14% 12% 8% 4% Error % Human A. thaliana
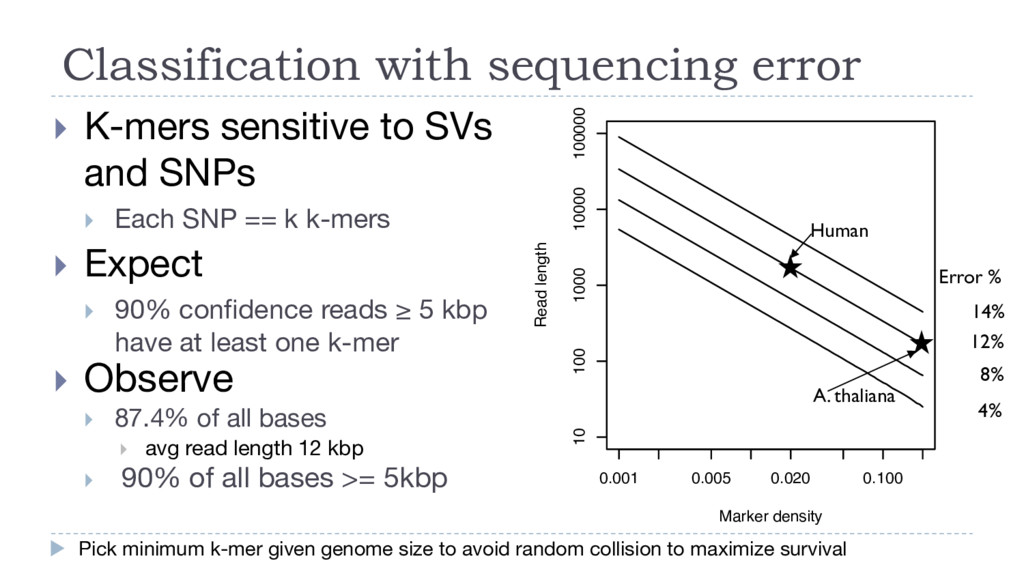
to avoid random collision to maximize survival } K-mers sensitive to SVs and SNPs } Each SNP == k k-mers 0.001 0.005 0.020 0.100 10 100 1000 10000 100000 Marker density Read length 14% 12% 8% 4% Error % Human A. thaliana } Expect } 90% confidence reads ≥ 5 kbp have at least one k-mer } Observe } 87.4% of all bases } avg read length 12 kbp } 90% of all bases >= 5kbp
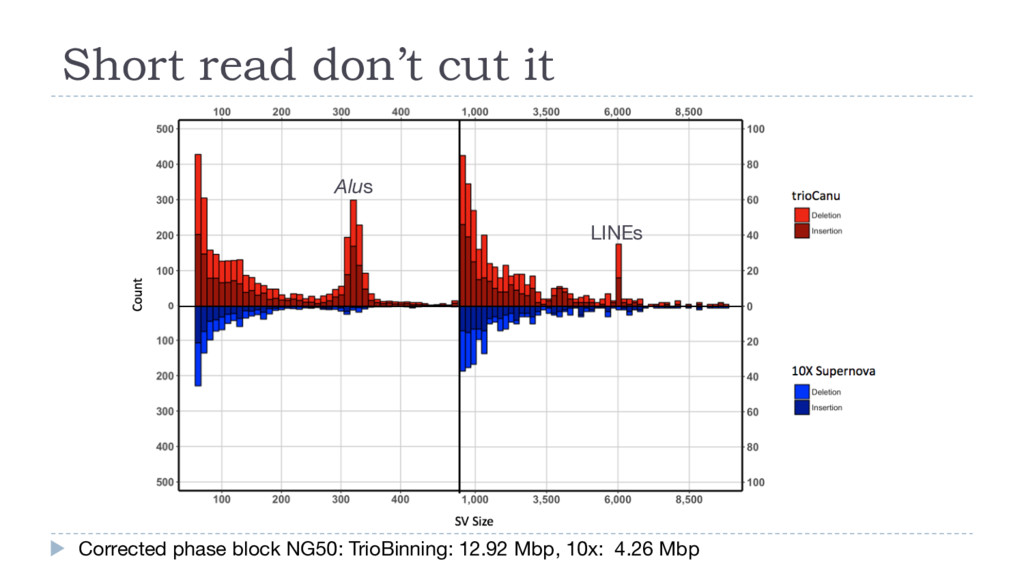
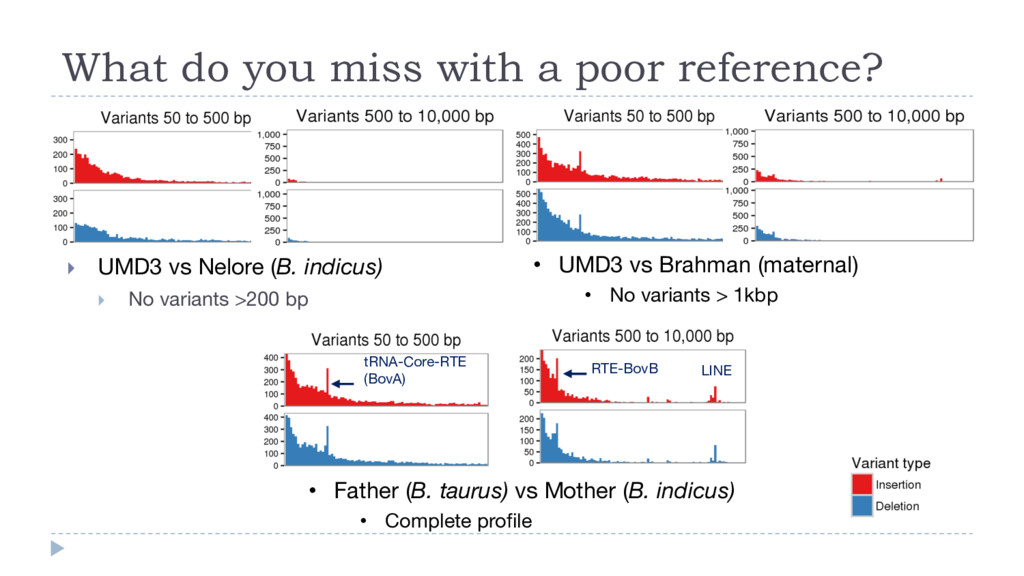
vs Nelore (B. indicus) } No variants >200 bp • UMD3 vs Brahman (maternal) • No variants > 1kbp • Father (B. taurus) vs Mother (B. indicus) • Complete profile LINE tRNA-Core-RTE (BovA) RTE-BovB
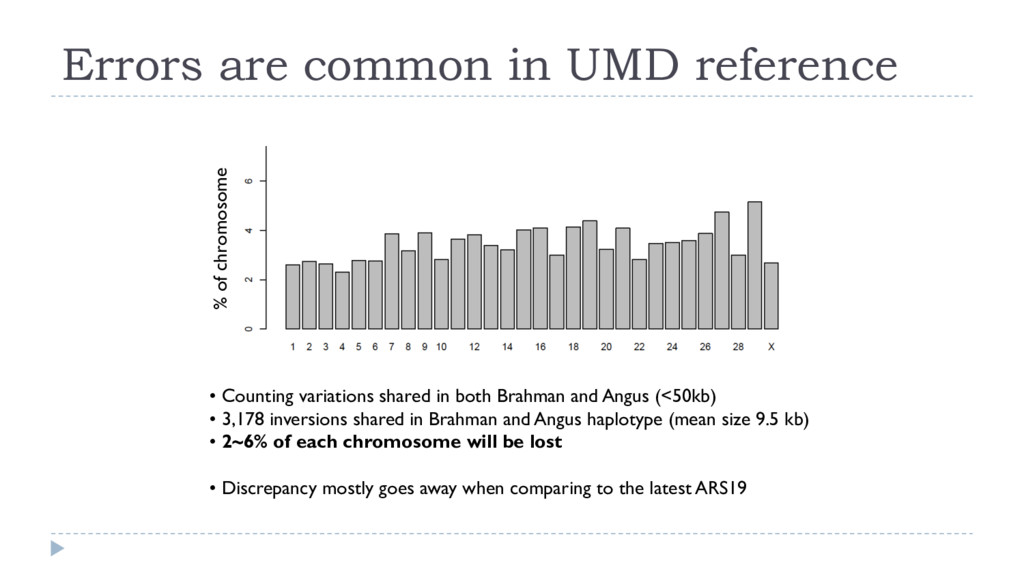
and Angus (<50kb) • 3,178 inversions shared in Brahman and Angus haplotype (mean size 9.5 kb) • 2~6% of each chromosome will be lost • Discrepancy mostly goes away when comparing to the latest ARS19 Errors are common in UMD reference
downloaded from BovineGenome.org } Genes in Angus assembly } 16,434 genes completely lifted over } 8,406 / 8,466 genes healed from gaps } Genes on chrY not lifted over } Genes in Brahman assembly } 18,105 genes completely lifted over } 9,366 / 9,401 genes healed from gaps } Heterozygosity (%) } Measuring SNPs, short INDELs, SVs when comparing Brahman and Angus assemblies } For each variation called in Brahman (D) and Angus (S); } Heterozygosity = 100 x { ∑max(D, S) / (1M + e ) } } where e = max(D, S) – min(D,S), extra sequence not in the 1M frame Brahman Angus 1 M e D S Measuring heterozygosity
Wrong strategy } Select most outbred individual along with parents to improve haplotype resolution } Get two full haplotypes phased across full genome } Greater continuity than assembling without trio information with sufficient coverage } Minimal additional cost of two Illumina libraries } Can also work with ancestral/survey data } Limited in regions of parent and child homozygosity (e.g. 0/1 genotype in all) } Trio approach cannot resolve unless spanned by reads ¨ Select more outbred individual ¨ Sequence with longer reads } Sequence/assembler agnostic } Polish/gap-fill as before using haplotype-assigned sequences } Combine with Hi-C to get haplotype resolved chromosomes A new strategy to generate references?
Rhie } Brian Walenz } Alexander Dilthey } Brian Ondov canu.readthedocs.io } Adam Phillippy } Sergey Koren } Brian Walenz } Konstantin Berlin } Jason Miller } Cow F1 collaborators } Tim Smith } John Williams } Sarah Kingan
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}