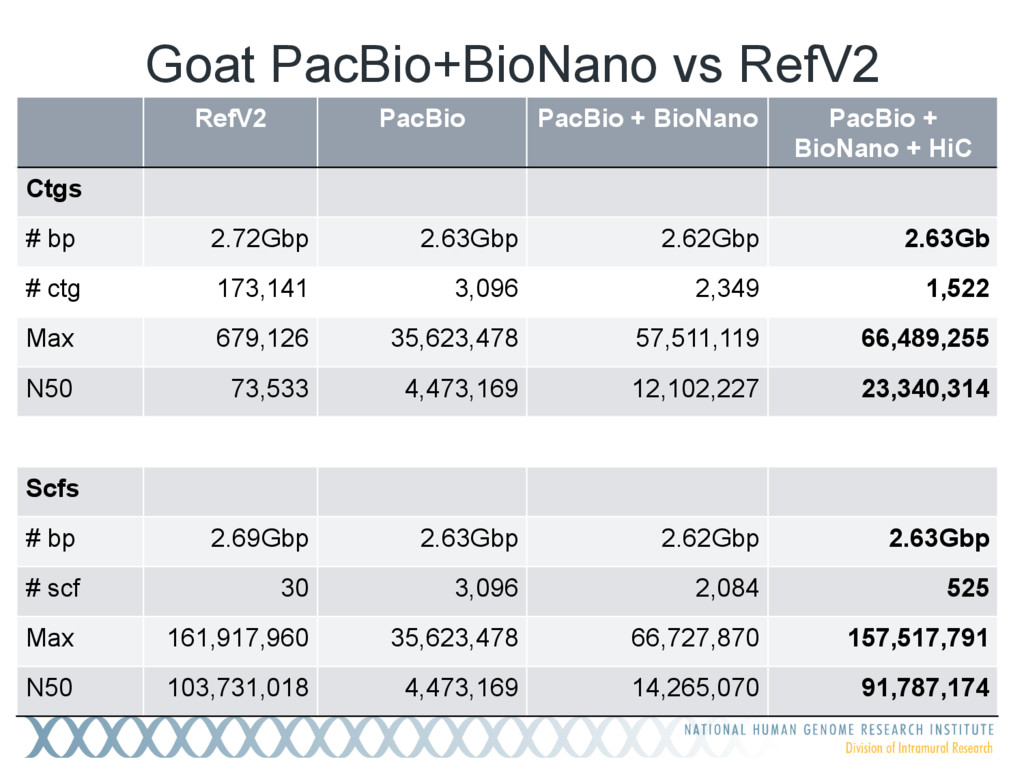
+ BioNano + HiC Ctgs # bp 2.72Gbp 2.63Gbp 2.62Gbp 2.63Gb # ctg 173,141 3,096 2,349 1,522 Max 679,126 35,623,478 57,511,119 66,489,255 N50 73,533 4,473,169 12,102,227 23,340,314 Scfs # bp 2.69Gbp 2.63Gbp 2.62Gbp 2.63Gbp # scf 30 3,096 2,084 525 Max 161,917,960 35,623,478 66,727,870 157,517,791 N50 103,731,018 4,473,169 14,265,070 91,787,174
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}