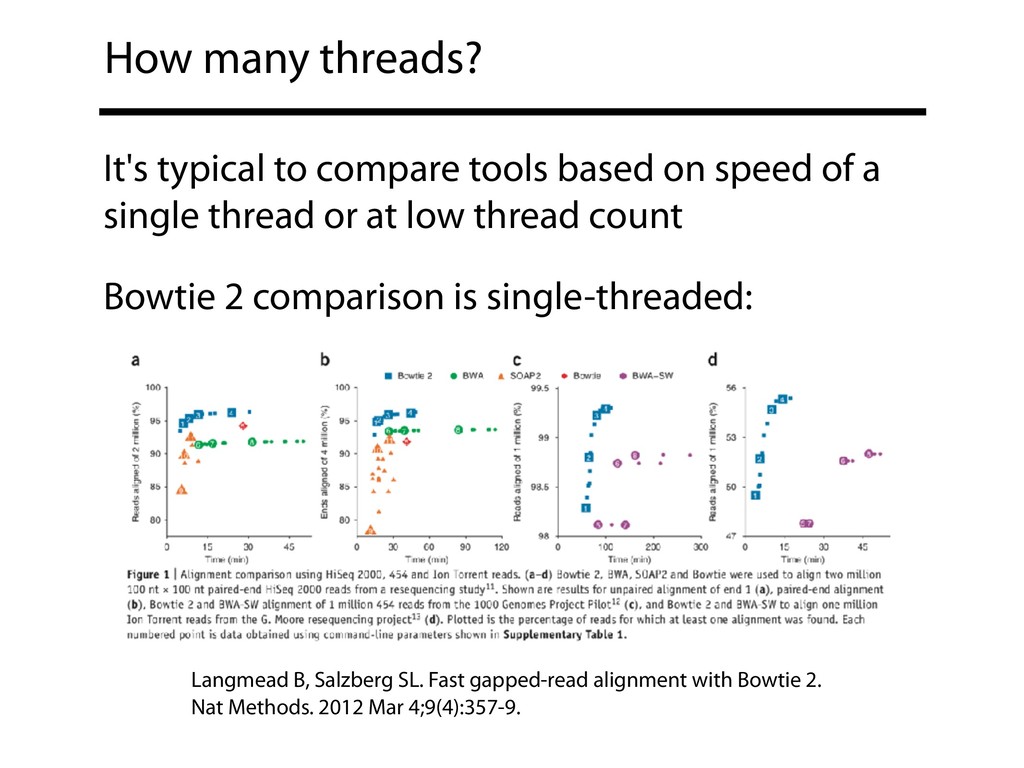
speed of a single thread or at low thread count Bowtie 2 comparison is single-threaded: Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012 Mar 4;9(4):357-9.
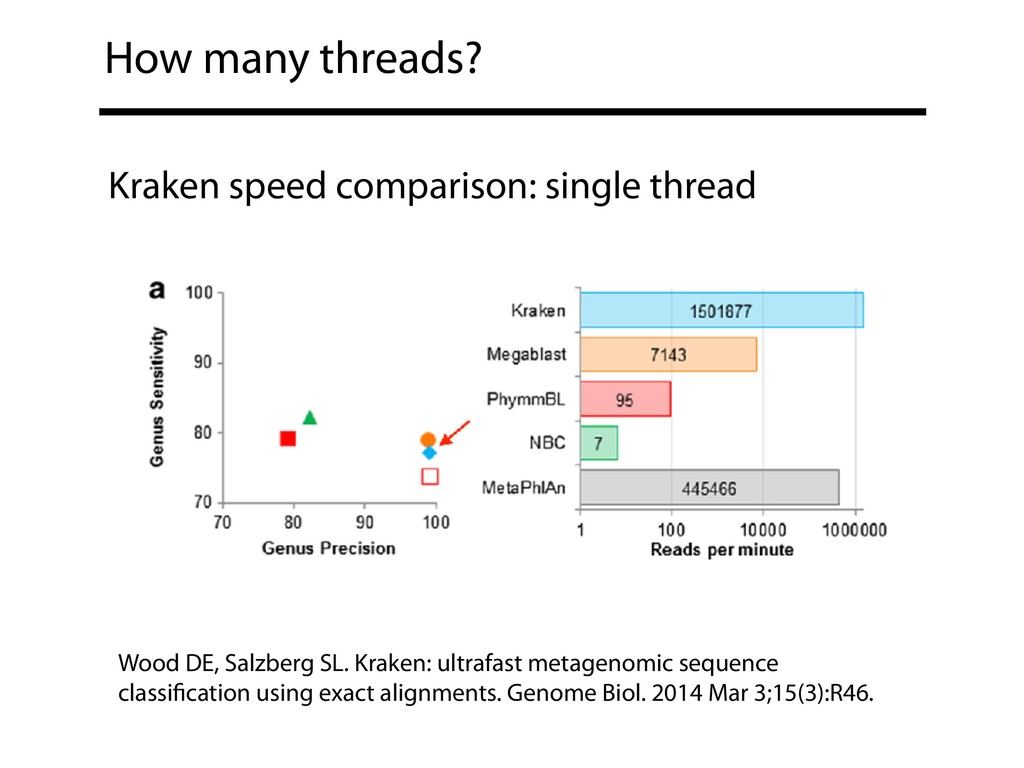
up to 8 threads Ounit R, Lonardi S. Higher classification sensitivity of short metagenomic reads with CLARK-S. Bioinformatics. 2016 Dec 15;32(24):3823-3825.
A performance comparison of data and memory allocation strategies for sequence aligners on NUMA architectures. Cluster Computing, 20(3), 1909-1924. Investigations at higher thread counts tend to be done by computer scientists; up to 64 here:
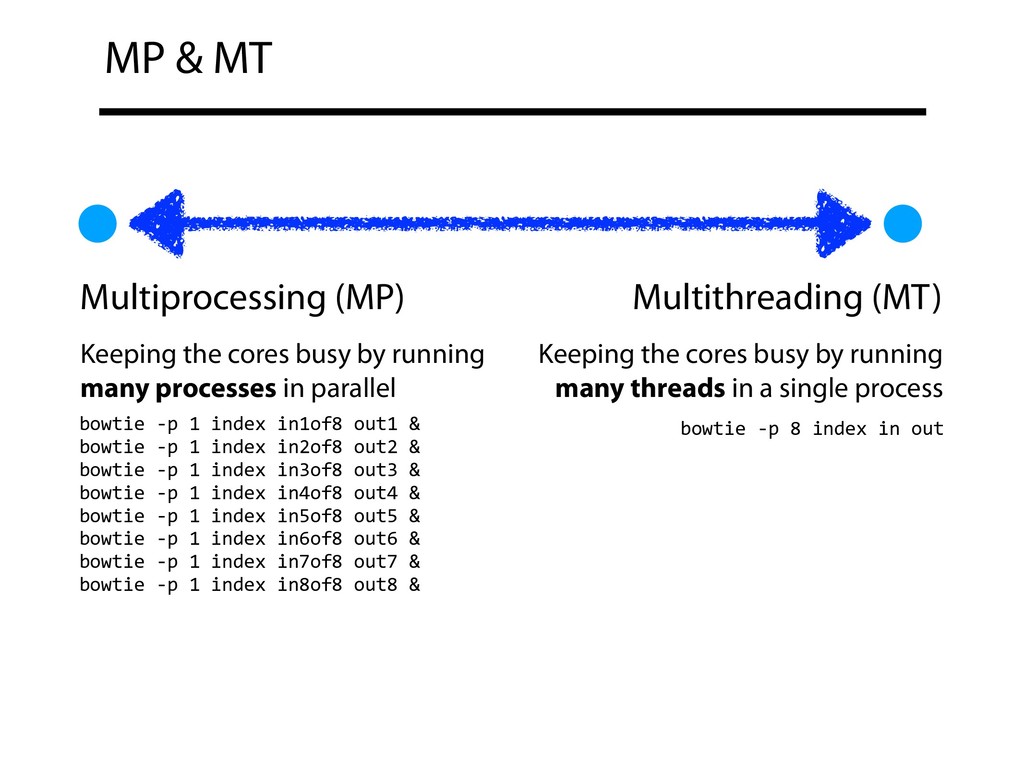
busy by running many processes in parallel Keeping the cores busy by running many threads in a single process bowtie -p 1 index in1of8 out1 & bowtie -p 1 index in2of8 out2 & bowtie -p 1 index in3of8 out3 & bowtie -p 1 index in4of8 out4 & bowtie -p 1 index in5of8 out5 & bowtie -p 1 index in6of8 out6 & bowtie -p 1 index in7of8 out7 & bowtie -p 1 index in8of8 out8 & bowtie -p 8 index in out
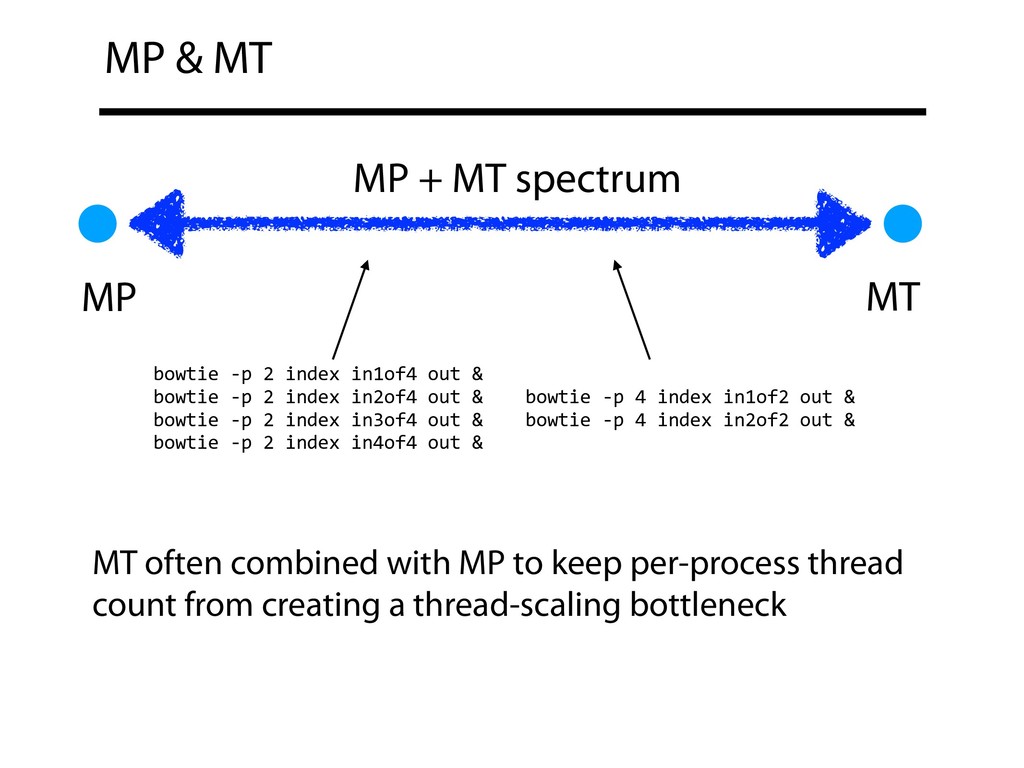
bowtie -p 4 index in2of2 out & MP MT bowtie -p 2 index in1of4 out & bowtie -p 2 index in2of4 out & bowtie -p 2 index in3of4 out & bowtie -p 2 index in4of4 out & MT often combined with MP to keep per-process thread count from creating a thread-scaling bottleneck MP + MT spectrum
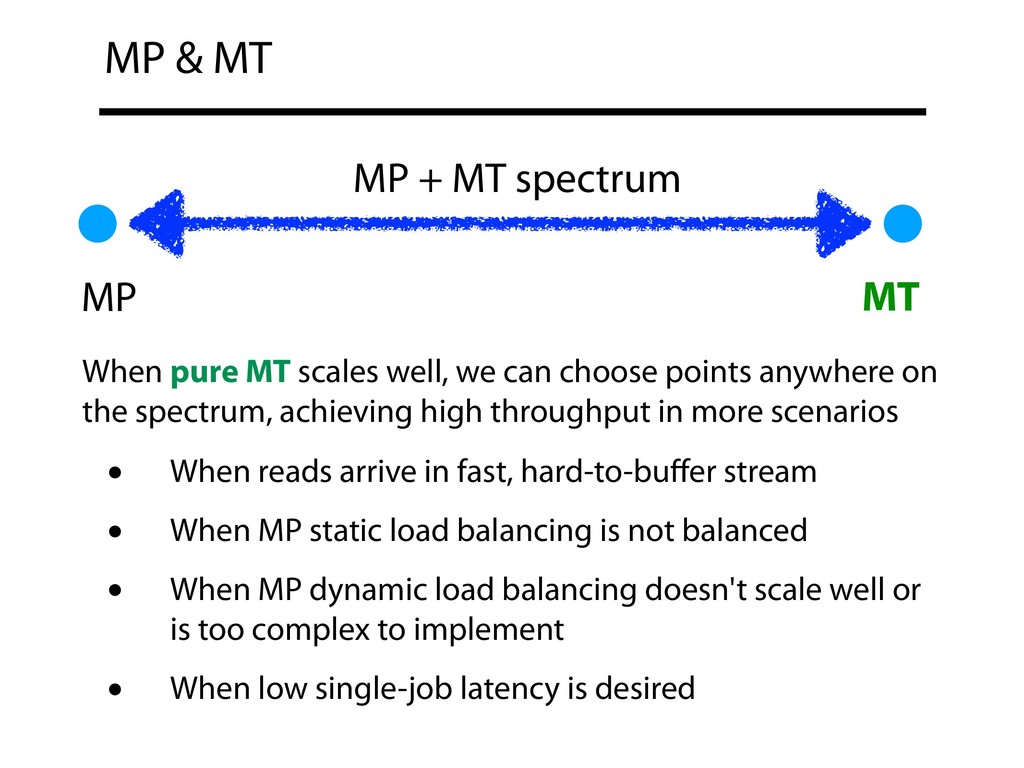
on the spectrum, achieving high throughput in more scenarios • When reads arrive in fast, hard-to-buffer stream • When MP static load balancing is not balanced • When MP dynamic load balancing doesn't scale well or is too complex to implement • When low single-job latency is desired MP MT MP & MT MP + MT spectrum
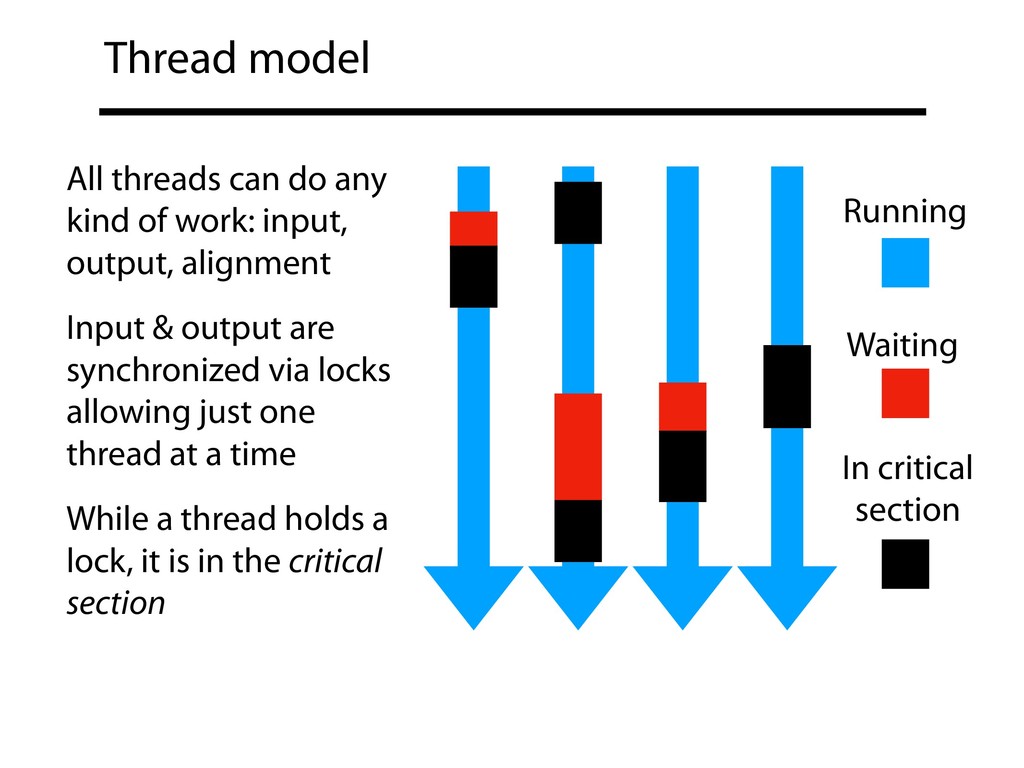
input, output, alignment Input & output are synchronized via locks allowing just one thread at a time While a thread holds a lock, it is in the critical section Running Waiting In critical section
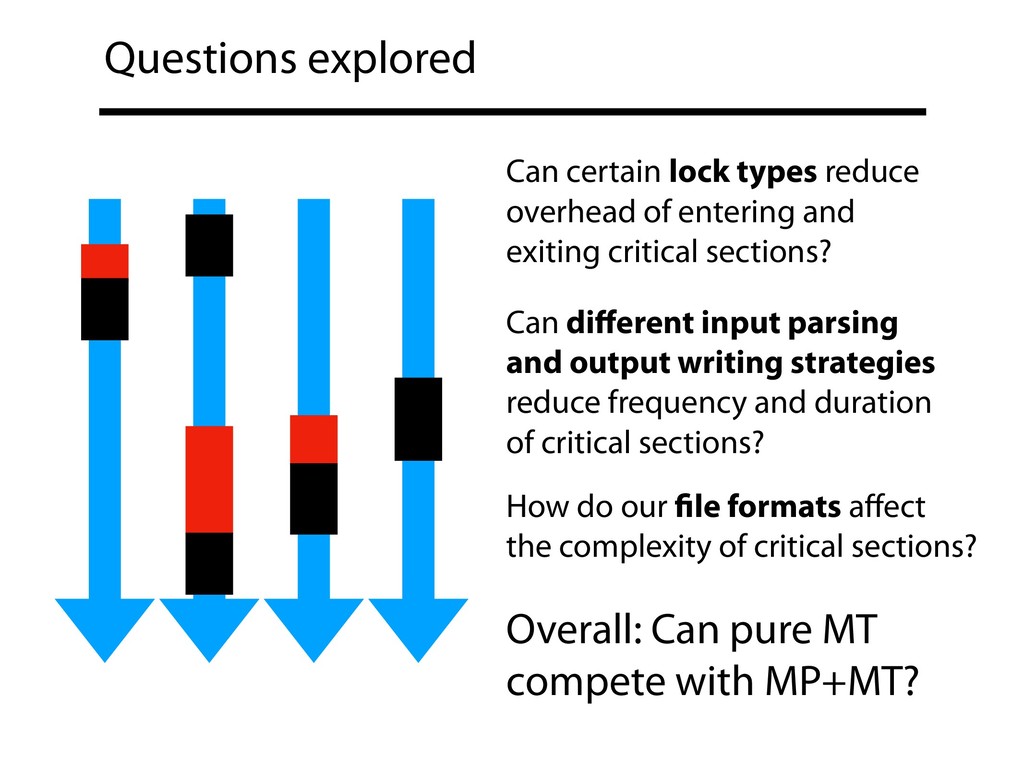
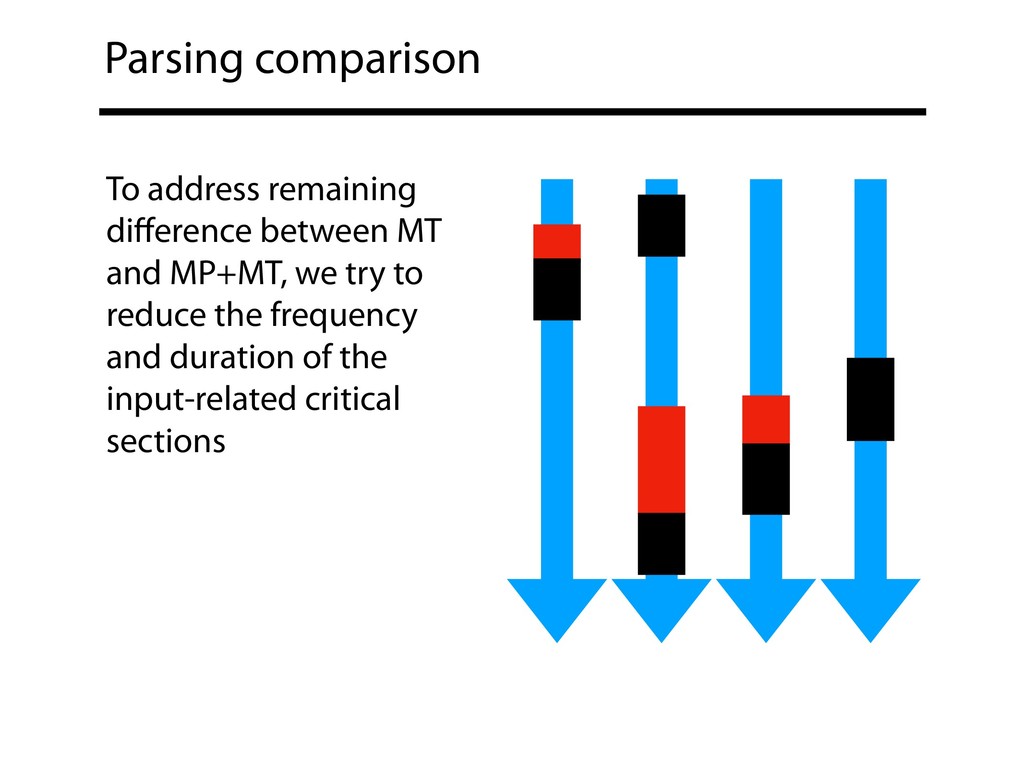
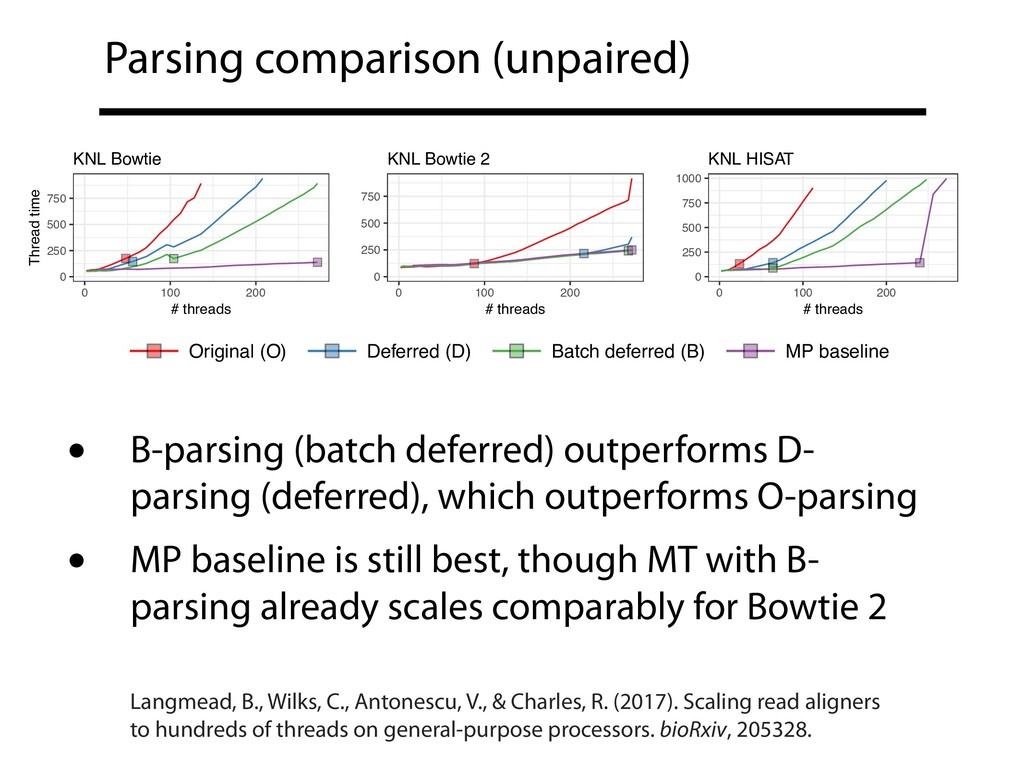
and exiting critical sections? Can different input parsing and output writing strategies reduce frequency and duration of critical sections? Overall: Can pure MT compete with MP+MT? How do our file formats affect the complexity of critical sections?
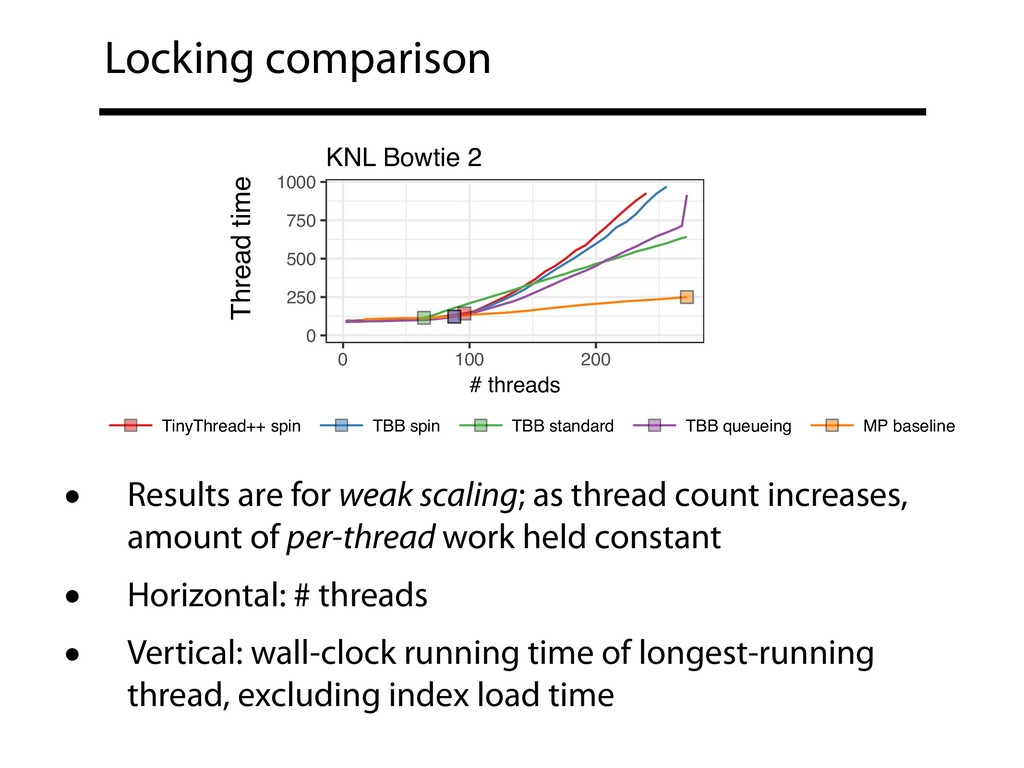
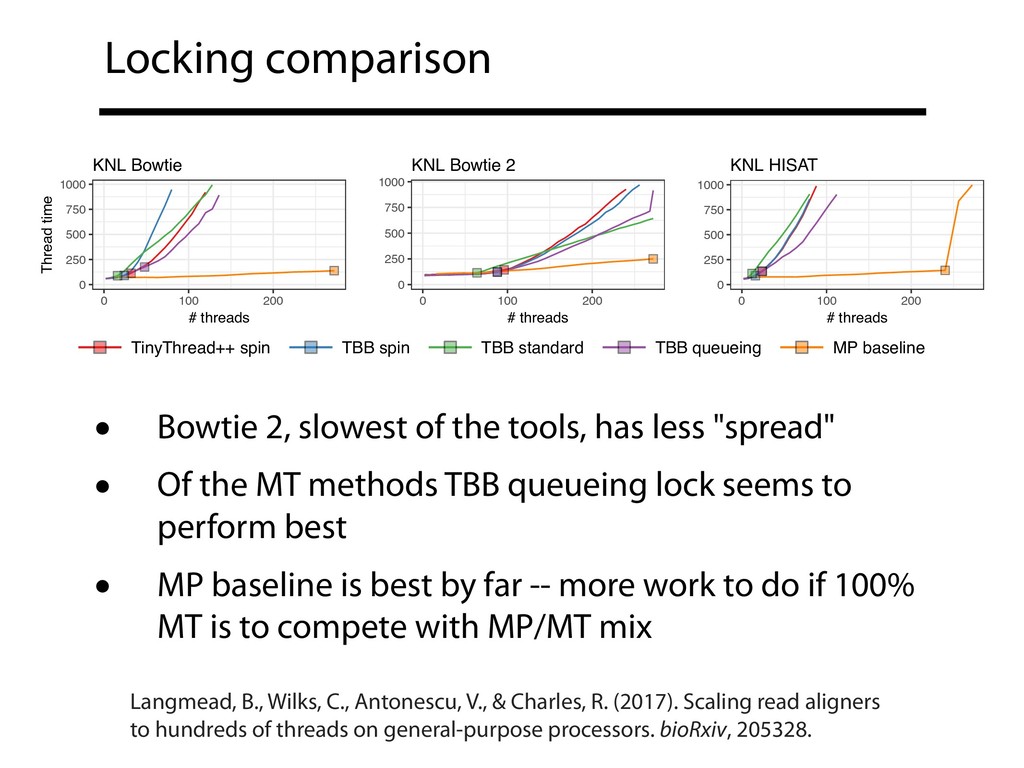
• All used here for (unspliced) DNA alignment only; HISAT's spliced alignment disabled • All code changes are in publicly-available branches (see preprint Supplementary Note 1) • Intel Thread Building Blocks (TBB) 2017 Update 5 Langmead, B., Wilks, C., Antonescu, V., & Charles, R. (2017). Scaling read aligners to hundreds of threads on general-purpose processors. bioRxiv, 205328.
with 68 cores, 96GB of DDR RAM, and 16GB of high speed MCDRAM • 1,736 Intel Xeon Skylake nodes, each with 48 cores and 192GB of RAM https://www.tacc.utexas.edu/systems/stampede2 At Texas Advanced Computing Center (TACC)
HiSeq 2000 100 x 100 paired-end sequencing: • Platinum Genomes (ERR194147) • 1000 Genomes Project (SRR069520) • Rustagi et al, low-coverage whole genome DNA sequencing of South Asian (SRR3947551) Aligned to hg38 Number of input reads per experiment tuned so 1 thread takes ~1 minute
spinning with a normal memory read • Atomic op treated like a write by cache coherence infrastructure • Many threads spinning at once leads to flood of cache coherence messages, clogging bus Details in preprint Langmead, B., Wilks, C., Antonescu, V., & Charles, R. (2017). Scaling read aligners to hundreds of threads on general-purpose processors. bioRxiv, 205328.
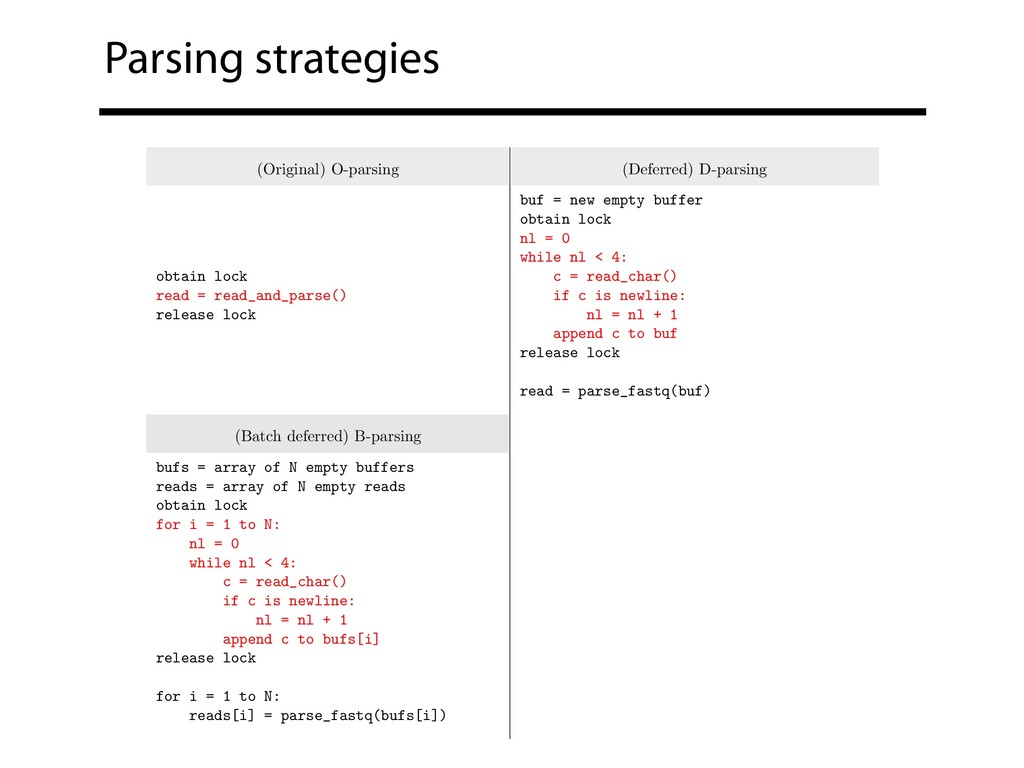
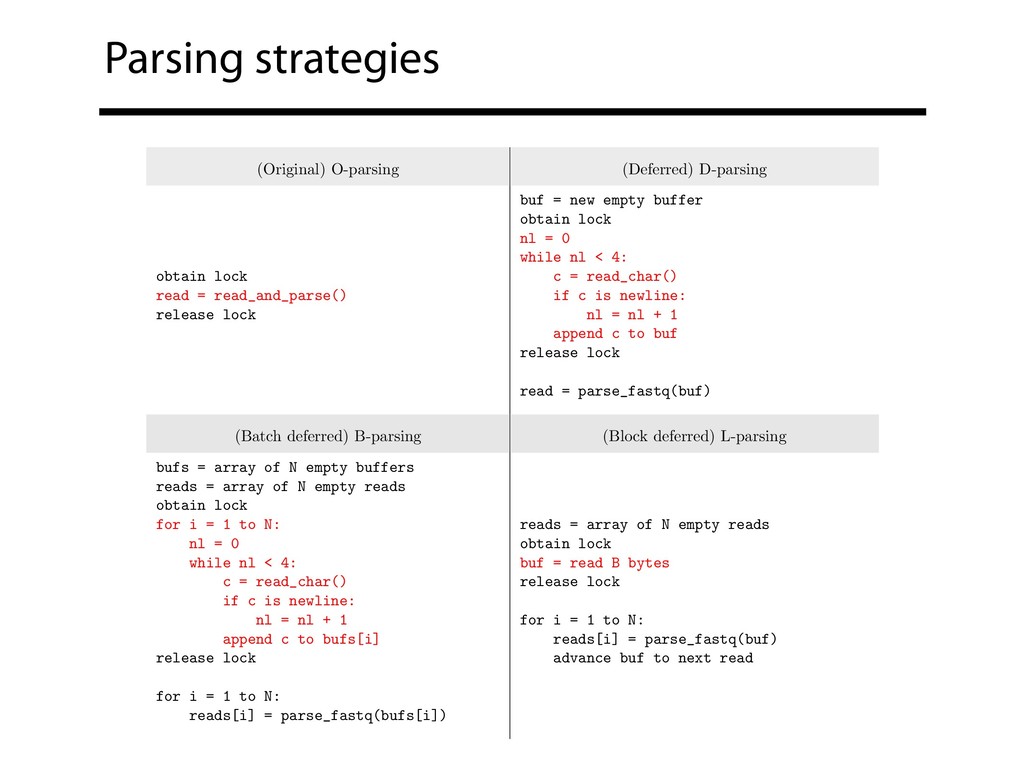
read_and_parse() release lock buf = new empty buffer obtain lock nl = 0 while nl < 4: c = read_char() if c is newline: nl = nl + 1 append c to buf release lock read = parse_fastq(buf) (Batch deferred) B-parsing (Block deferred) L-parsing bufs = array of N empty buffers reads = array of N empty reads obtain lock for i = 1 to N: nl = 0 while nl < 4: c = read_char() if c is newline: nl = nl + 1 append c to bufs[i] release lock for i = 1 to N: reads[i] = parse_fastq(bufs[i]) reads = array of N empty reads obtain lock buf = read B bytes release lock for i = 1 to N: reads[i] = parse_fastq(buf) advance buf to next read
read_and_parse() release lock buf = new empty buffer obtain lock nl = 0 while nl < 4: c = read_char() if c is newline: nl = nl + 1 append c to buf release lock read = parse_fastq(buf) (Batch deferred) B-parsing (Block deferred) L-parsing bufs = array of N empty buffers reads = array of N empty reads obtain lock for i = 1 to N: nl = 0 while nl < 4: c = read_char() if c is newline: nl = nl + 1 append c to bufs[i] release lock for i = 1 to N: reads[i] = parse_fastq(bufs[i]) reads = array of N empty reads obtain lock buf = read B bytes release lock for i = 1 to N: reads[i] = parse_fastq(buf) advance buf to next read
read_and_parse() release lock buf = new empty buffer obtain lock nl = 0 while nl < 4: c = read_char() if c is newline: nl = nl + 1 append c to buf release lock read = parse_fastq(buf) (Batch deferred) B-parsing (Block deferred) L-parsing bufs = array of N empty buffers reads = array of N empty reads obtain lock for i = 1 to N: nl = 0 while nl < 4: c = read_char() if c is newline: nl = nl + 1 append c to bufs[i] release lock for i = 1 to N: reads[i] = parse_fastq(bufs[i]) reads = array of N empty reads obtain lock buf = read B bytes release lock for i = 1 to N: reads[i] = parse_fastq(buf) advance buf to next read
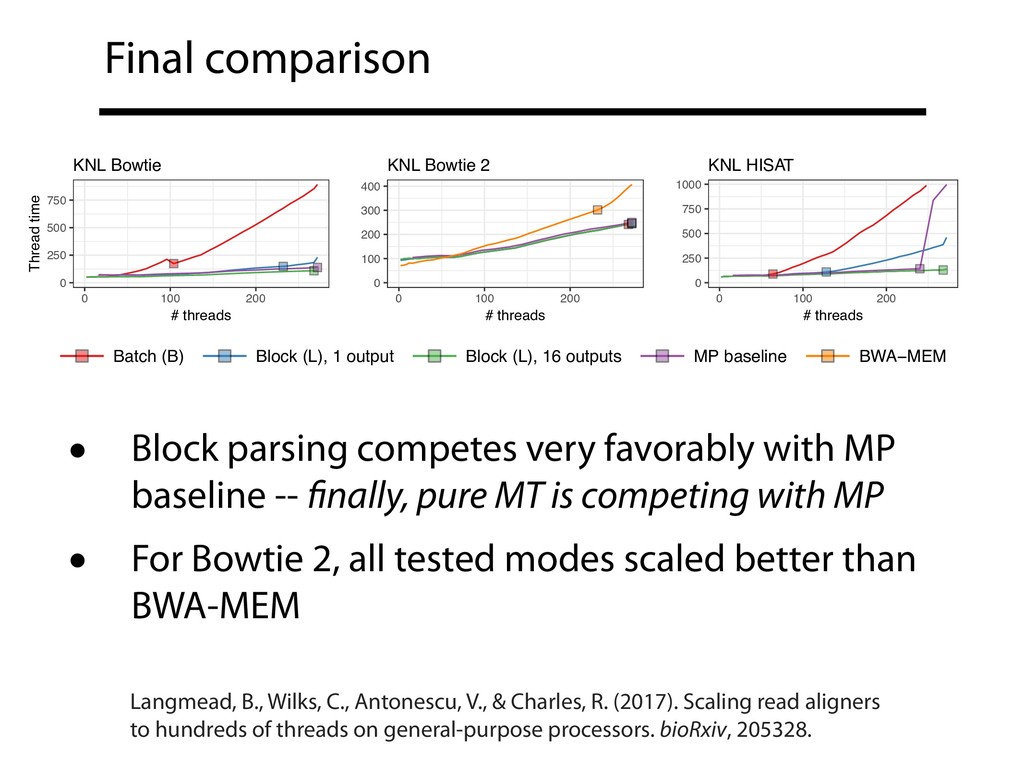
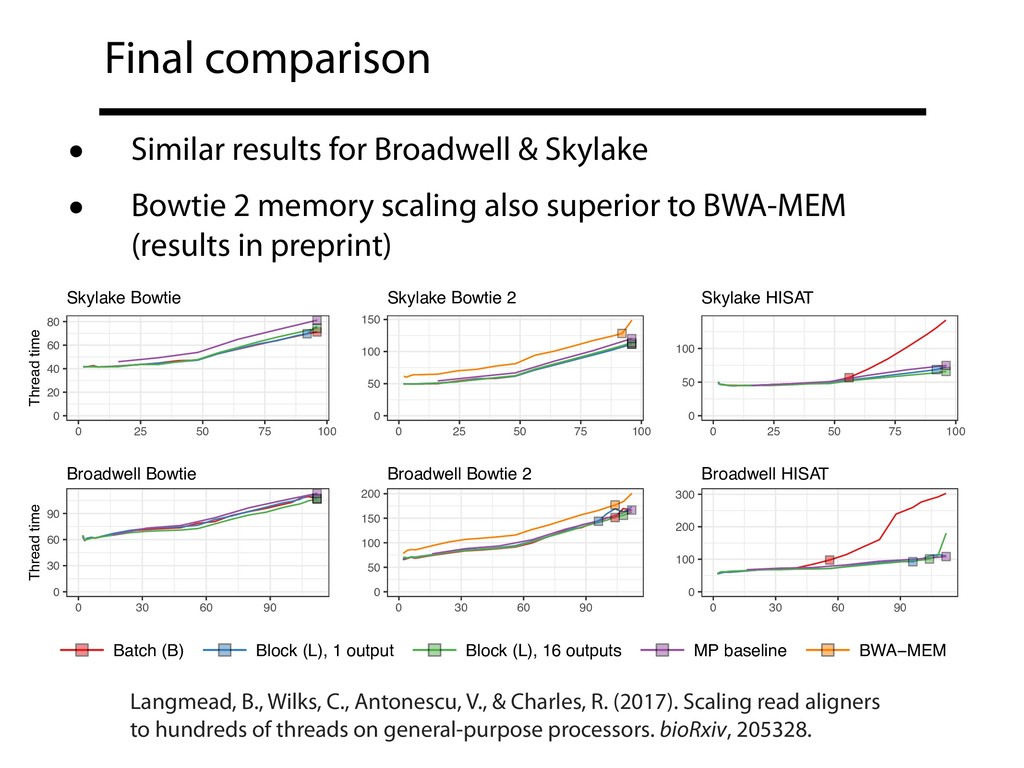
repos • See supplementary note 1 in preprint B-parsing with the TBB queueing lock is now the default in Bowtie & Bowtie 2 • Block (L) parsing can't be the default because typical FASTQ isn't padded Langmead, B., Wilks, C., Antonescu, V., & Charles, R. (2017). Scaling read aligners to hundreds of threads on general-purpose processors. bioRxiv, 205328.
well as MP+MT, unlocking full spectrum of MP/MT trade offs • Thread scaling and file formats are linked; our (least) favorite file formats could be better designed with an eye to thread scaling • Gains described here can generalize to other embarrassingly parallel software • MT scaling should be a first-class concern when describing and evaluating upstream genomics tools in literature
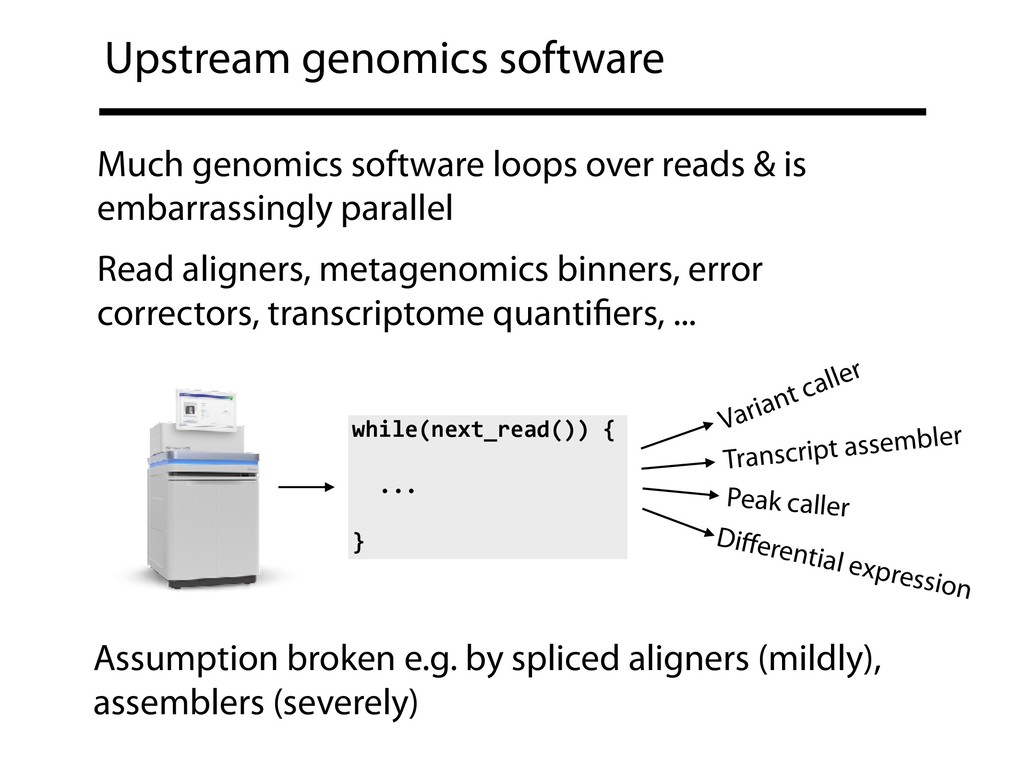
processors Ben Langmead1,2,*, Christopher Wilks1,2, Valentin Antonescu1, and Rone Charles1 1Department of Computer Science, Johns Hopkins University 2Center for Computational Biology, Johns Hopkins University *Correspondence to: [email protected] February 4, 2018 Abstract General-purpose processors can now contain many dozens of processor cores and support hundreds of simultaneous threads of execution. To make best use of these threads, genomics software must contend with new and subtle computer architecture issues. We discuss some of these and propose methods for improving thread scaling in tools that analyze each read in- dependently, such as read aligners. We implement these methods in new versions of Bowtie, Bowtie 2 and HISAT. We greatly improve thread scaling in many scenarios, including on the re- cent Intel Xeon Phi architecture. We also highlight how bottlenecks are exacerbated by variable- record-length file formats like FASTQ and suggest changes that enable superior scaling. 1 Introduction General-purpose processors are now capable of running hundreds of threads of execution simul- taneously in parallel. Intel’s Xeon Phi “Knight’s Landing” architecture supports 256–288 simul- taneous threads across 64–72 physical processor cores [1, 2]. With severe physical limits on clock speed [3], future architectures will likely support more simultaneous threads rather than faster in- Langmead, B., Wilks, C., Antonescu, V., & Charles, R. (2017). Scaling read aligners to hundreds of threads on general-purpose processors. bioRxiv, 205328.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}