## Density of states of conjugated polymers by tight binding
### Jarvist Moore Frost, Beth Rice, Jenny Nelson
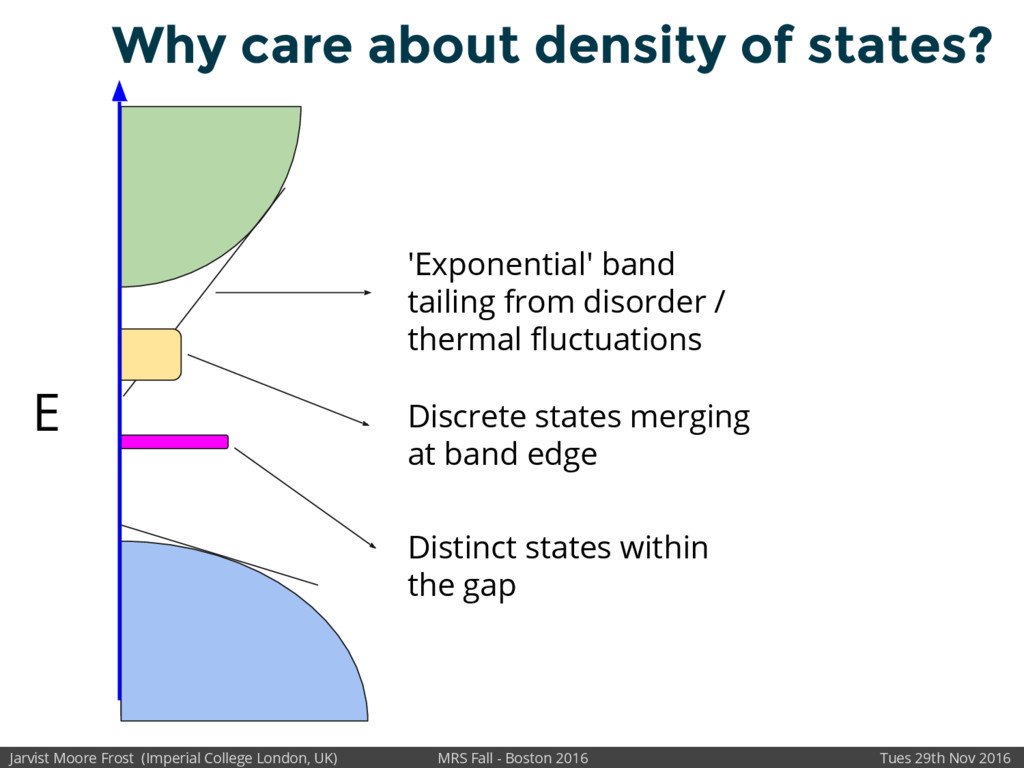
Organic electronic materials are highly spatially disordered. Resultant fluctuations in wavefunction overlap leads to band tailing in the electronic density of states. In turn this limits the charge carrier mobility. Methods to understand these relationships will enable the design of higher performance organic semiconductors.
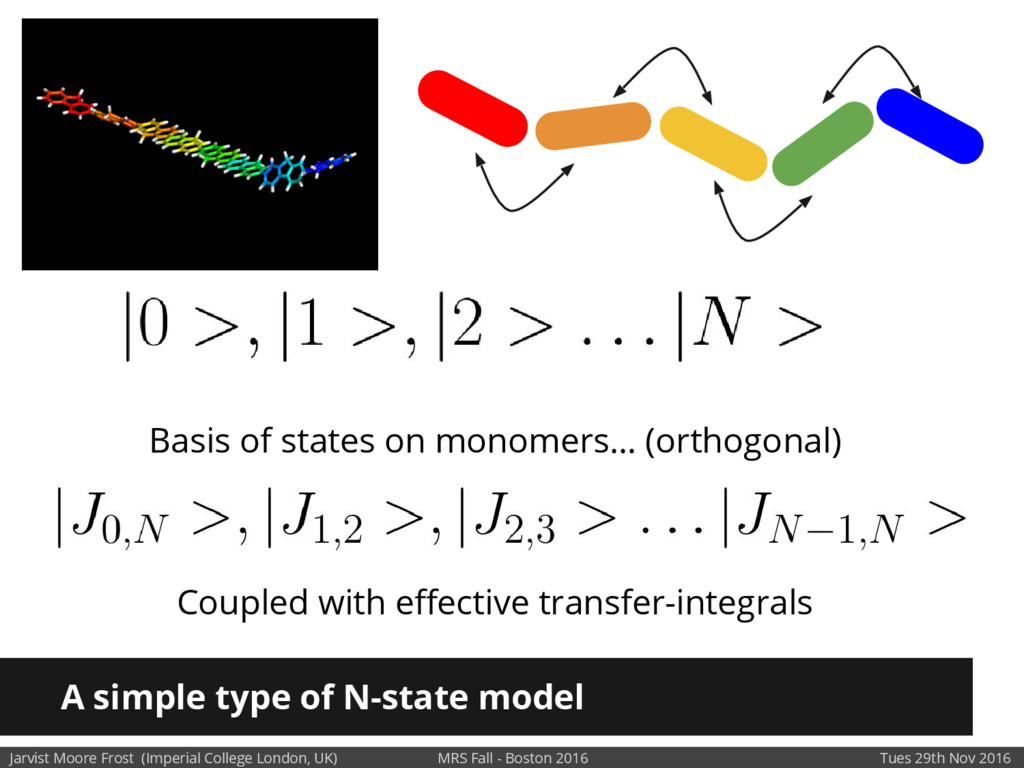
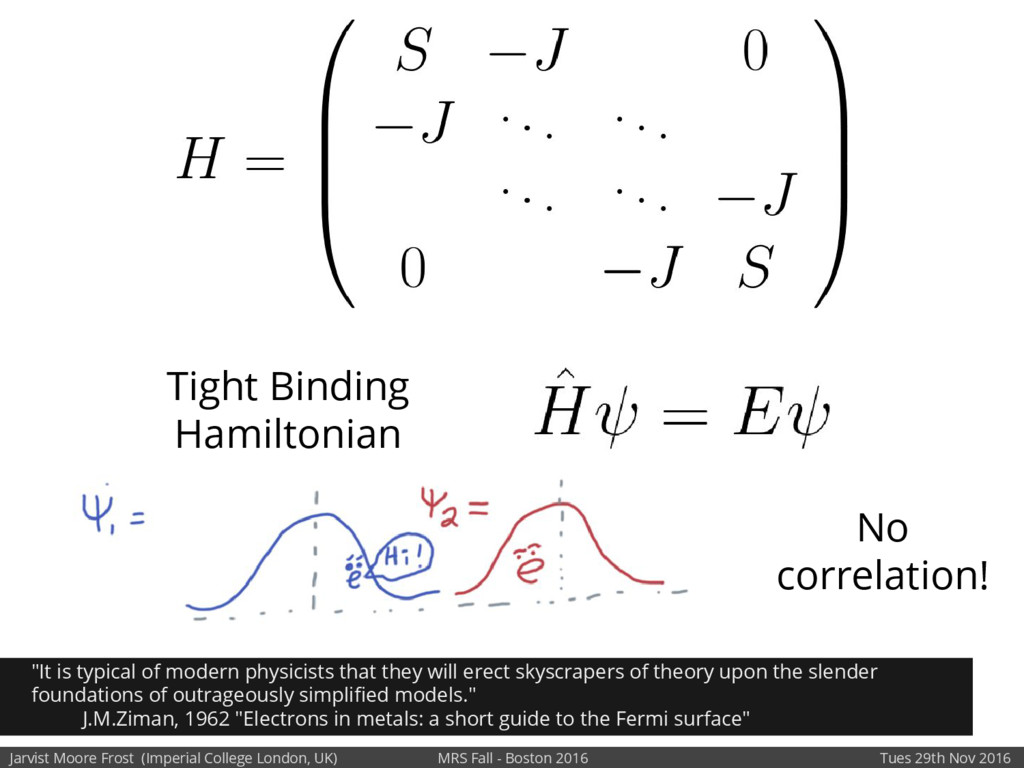
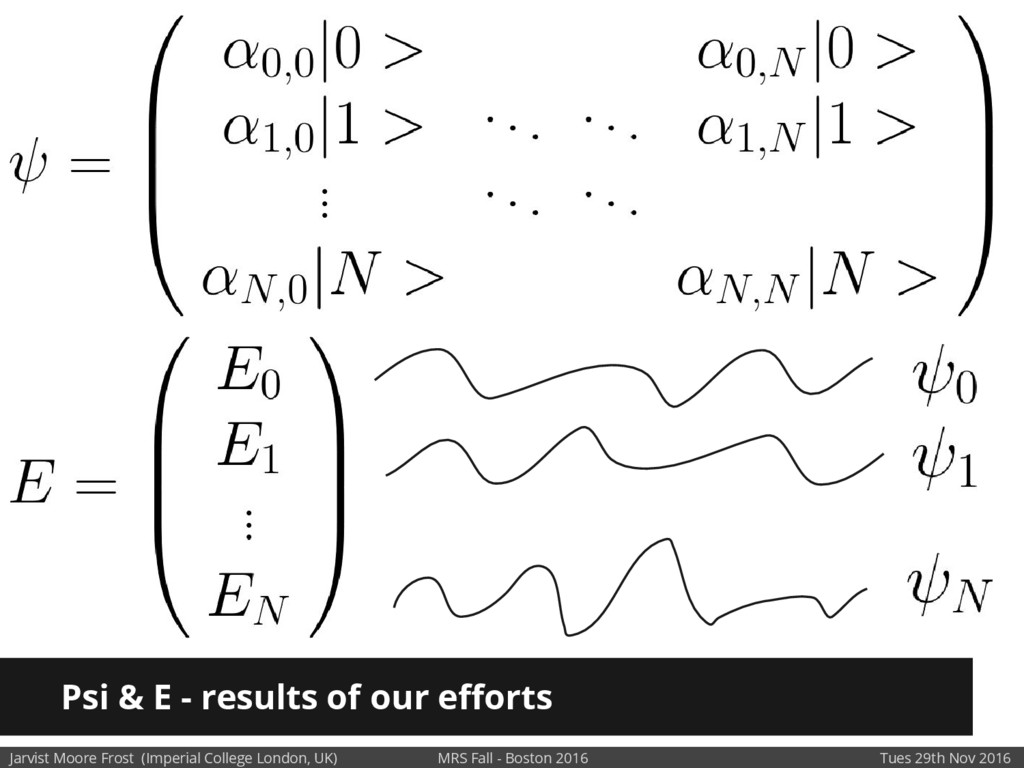
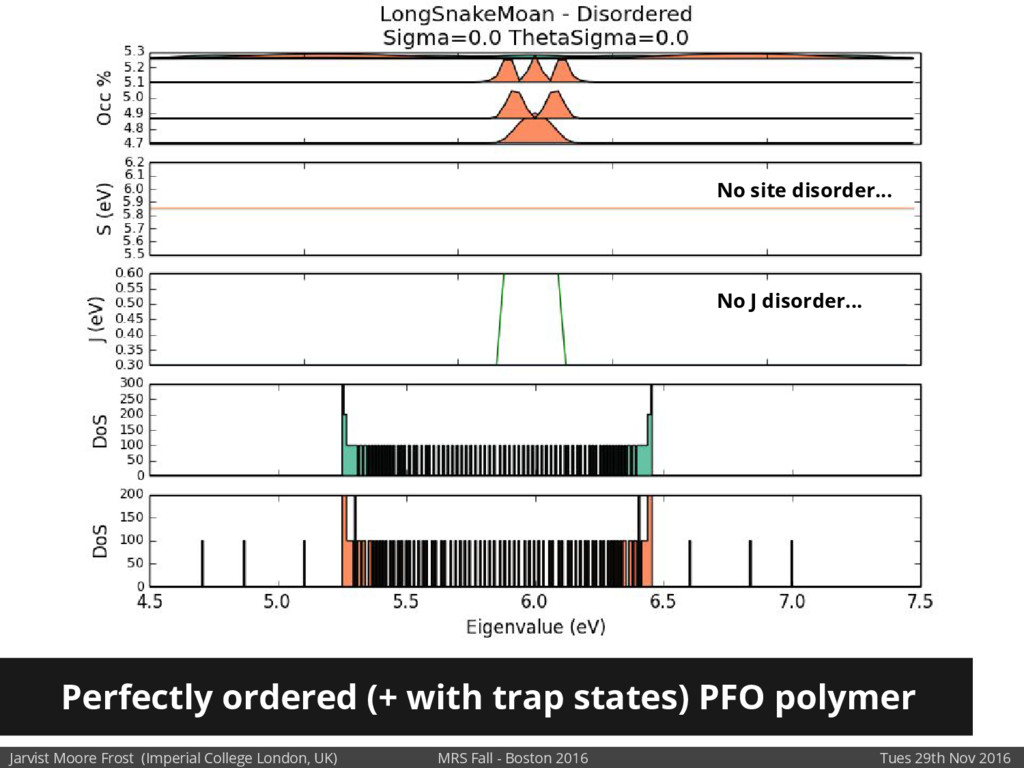
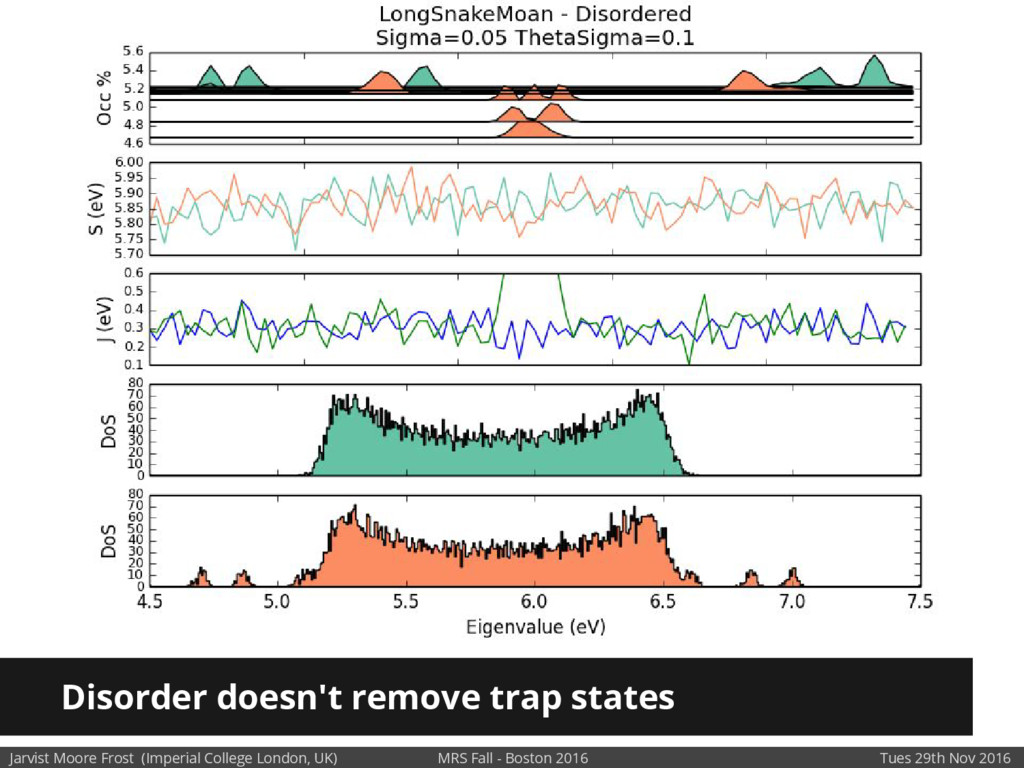
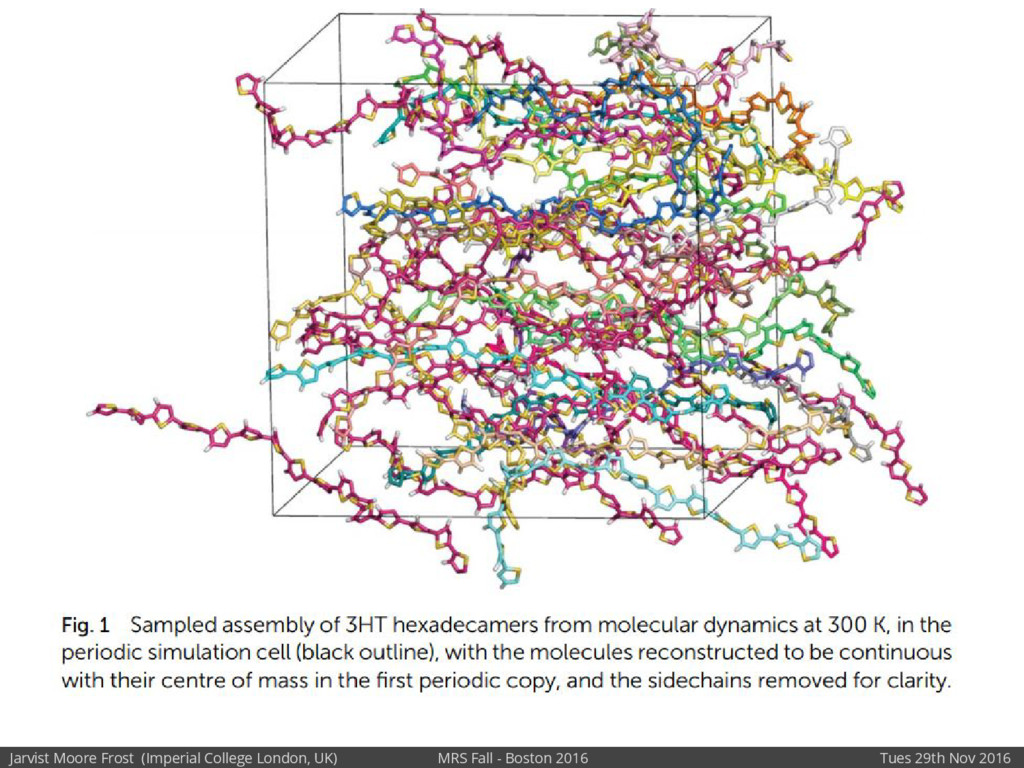
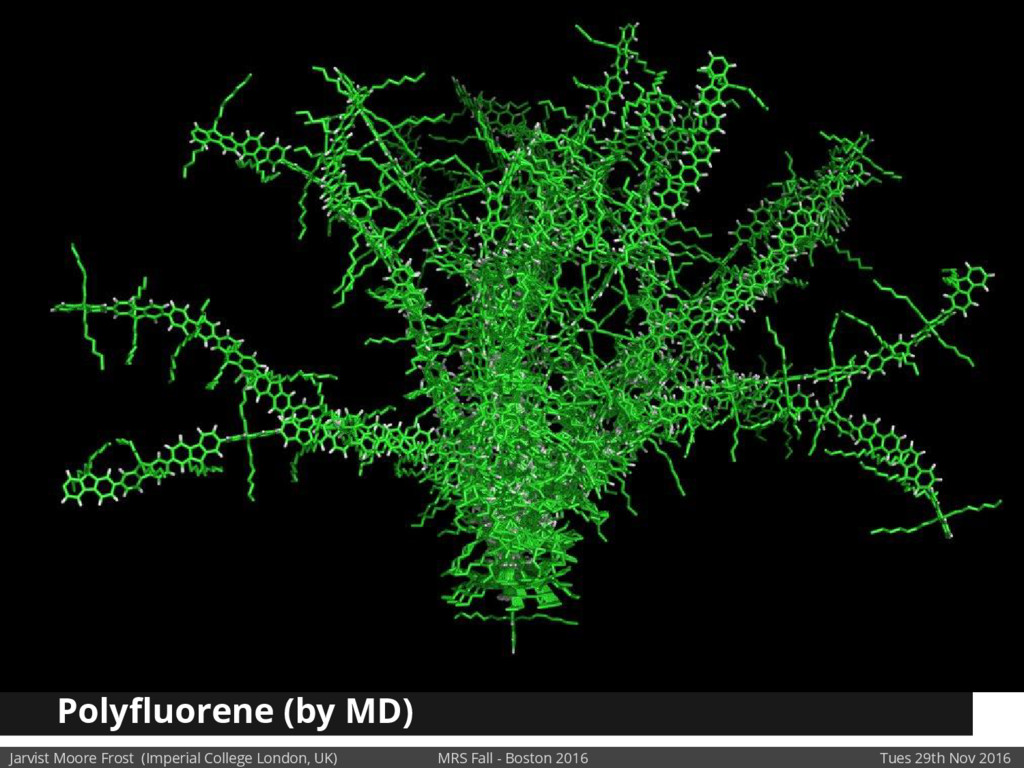
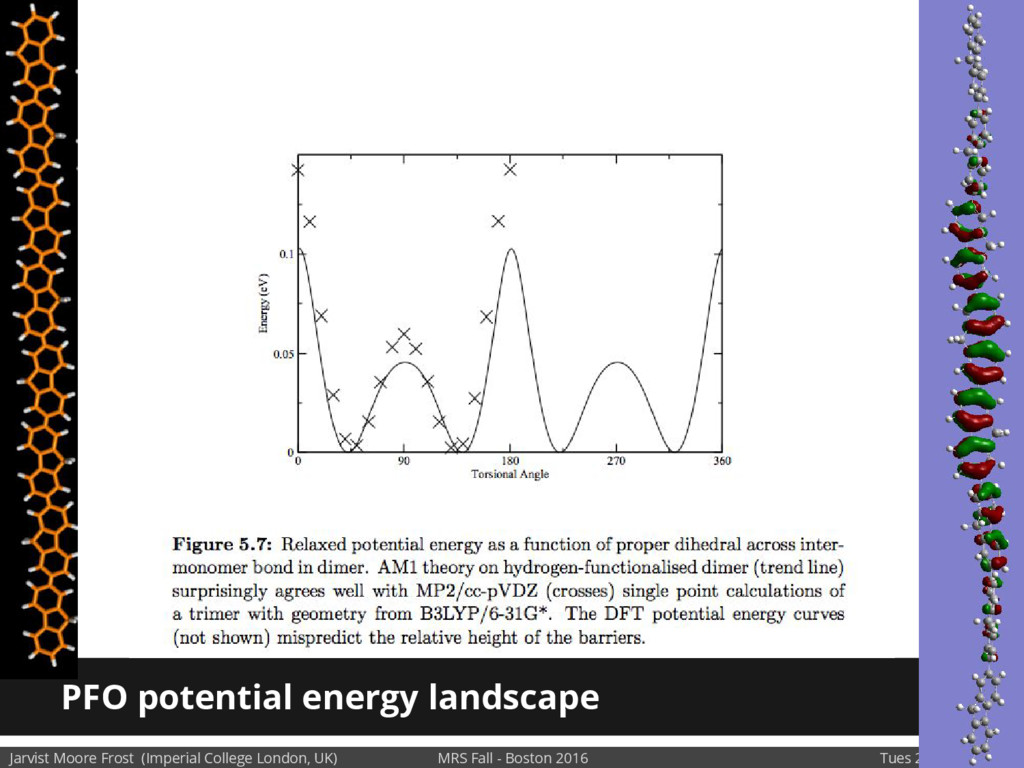
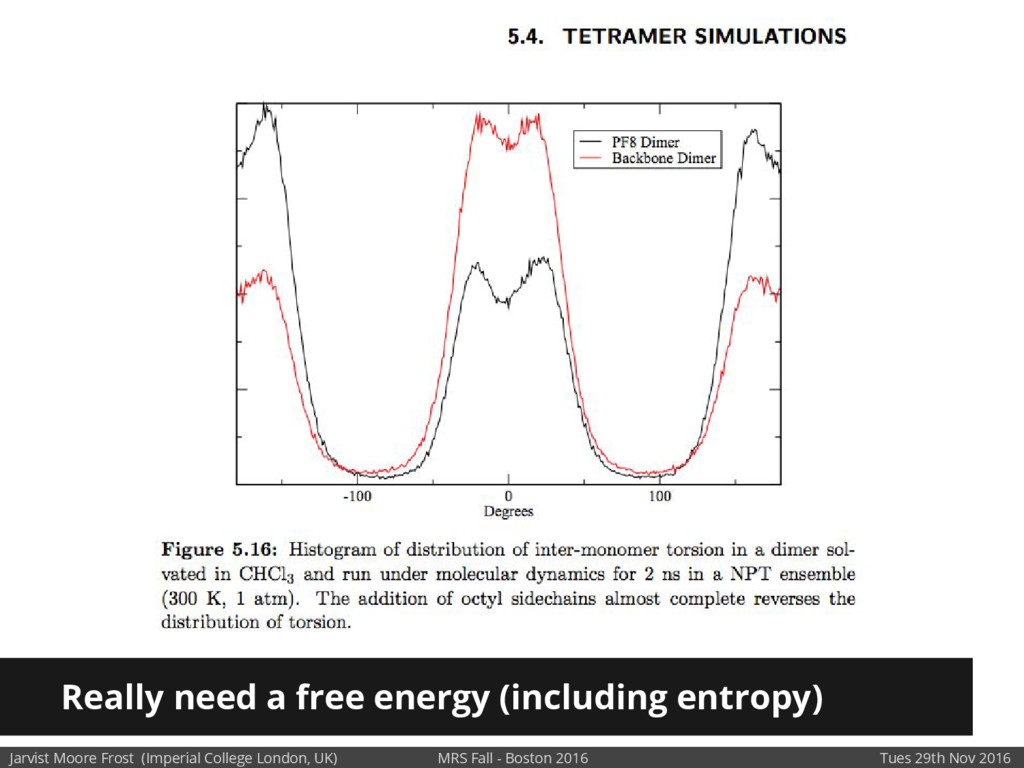
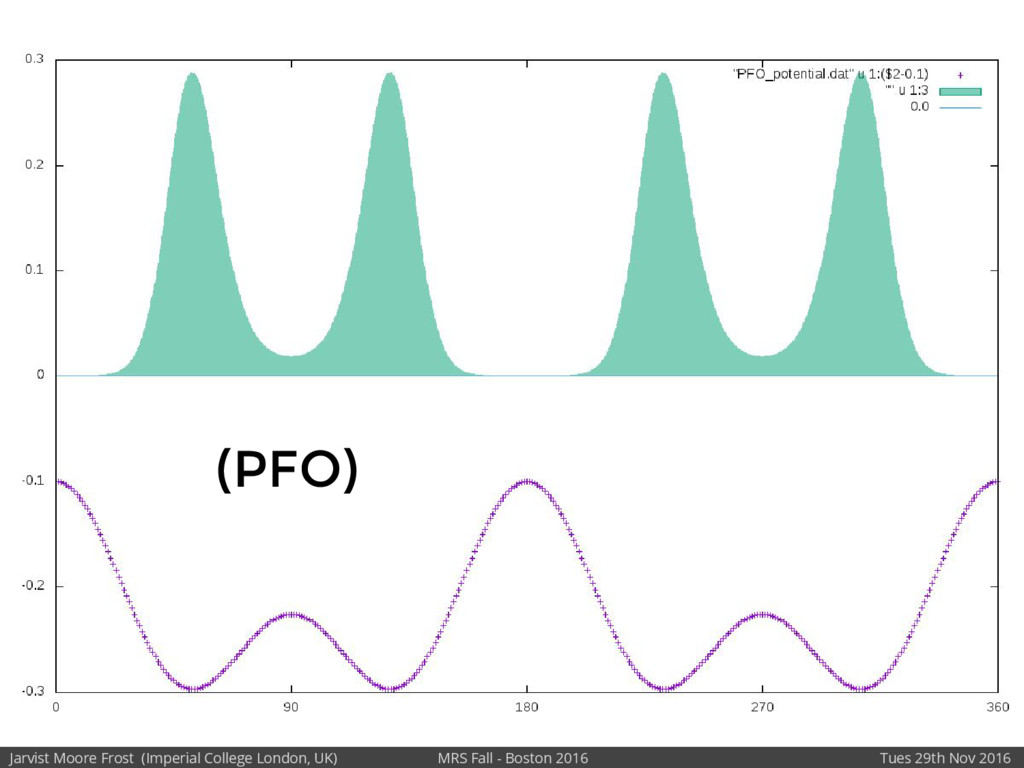
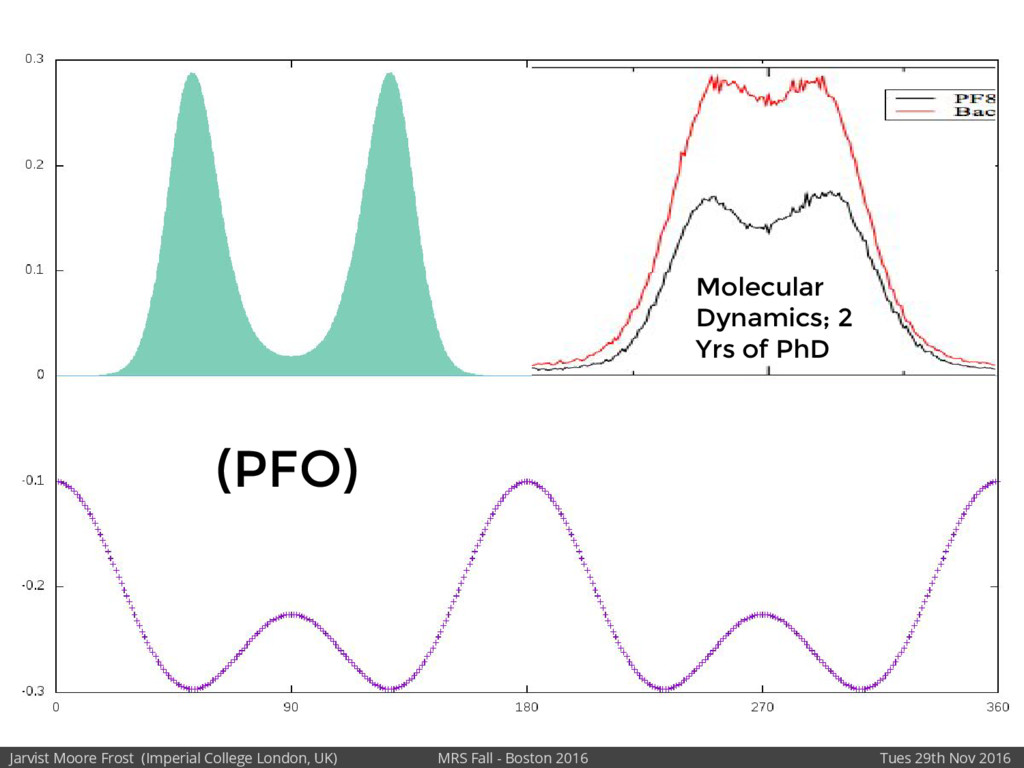
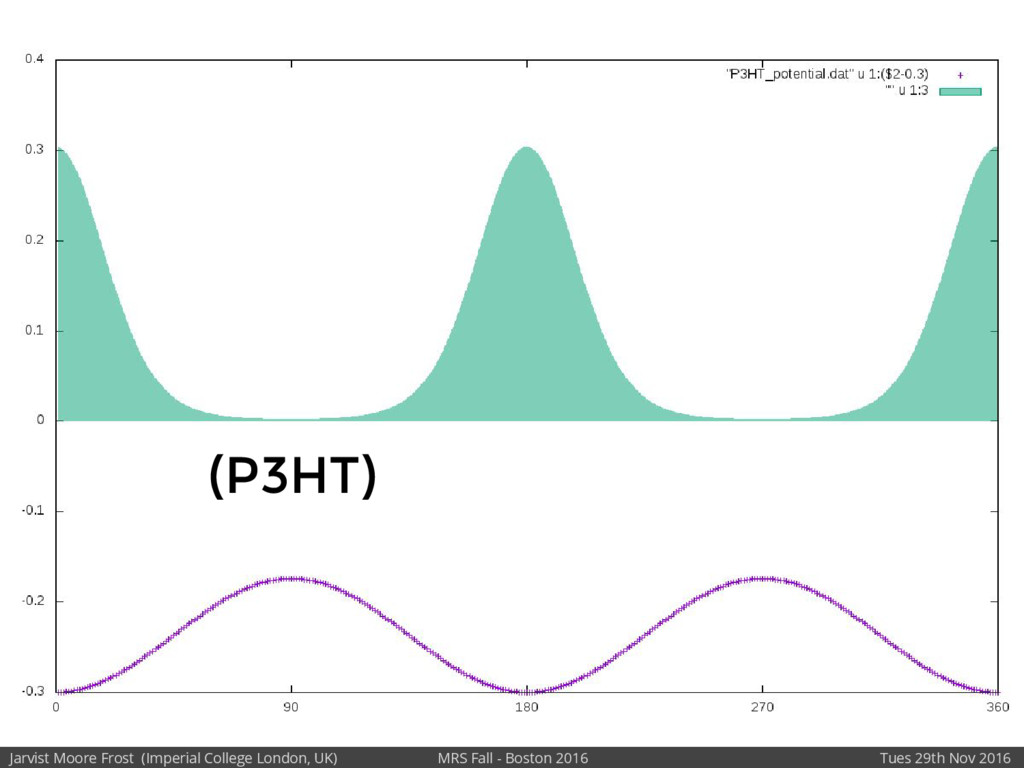
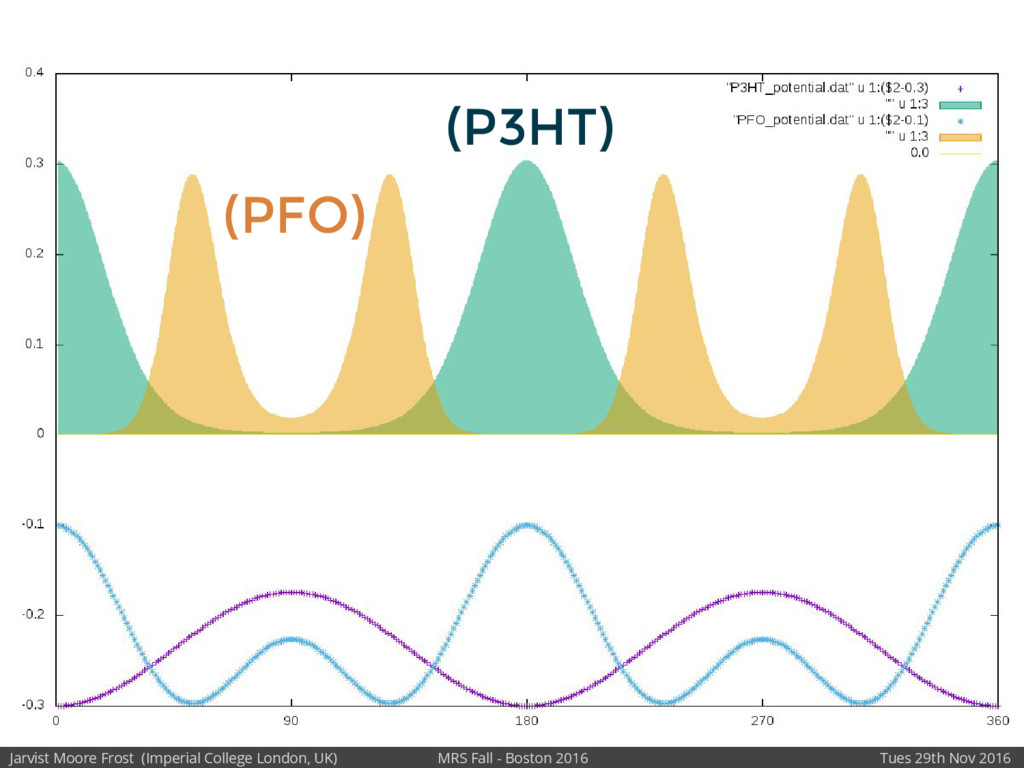
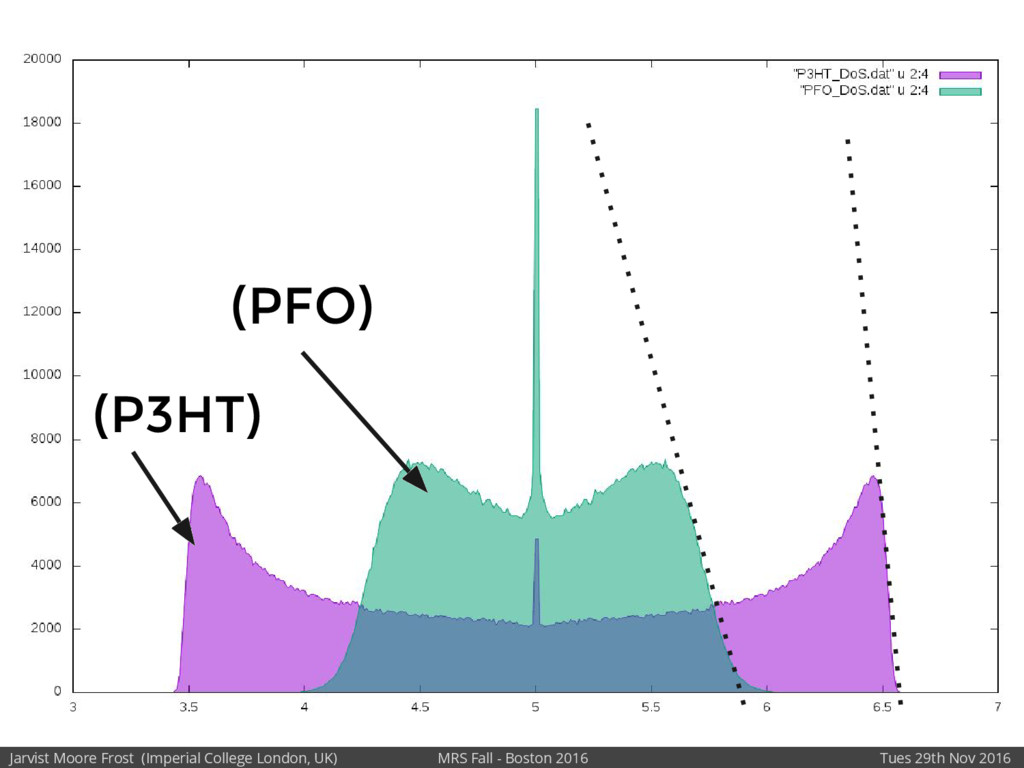
We will discuss the multi-scale simulation methods required to solve the electronic density of states of a conjugated polymer, focusing on amorphous P3HT[1]. Here we undertook atomistic molecular dynamics, calculated transfer integrals from frozen snapshots with the molecular orbital overlap method, and then solved a tight binding model, to have an entirely ab-initio prediction of the Urbach tail of charges. Our chief results were: inter-monomer torsional disorder dominates intra-chain disorder; and that the Urbach tail was composed of extremal configurations, and so required very large calculations to converge on a value.
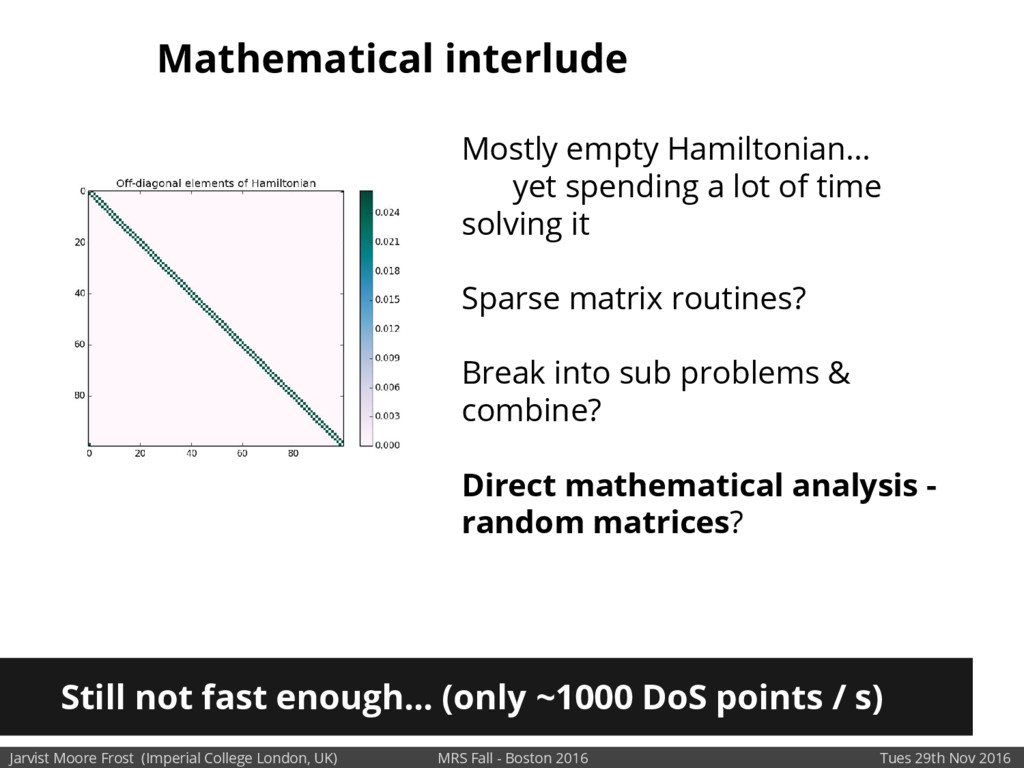
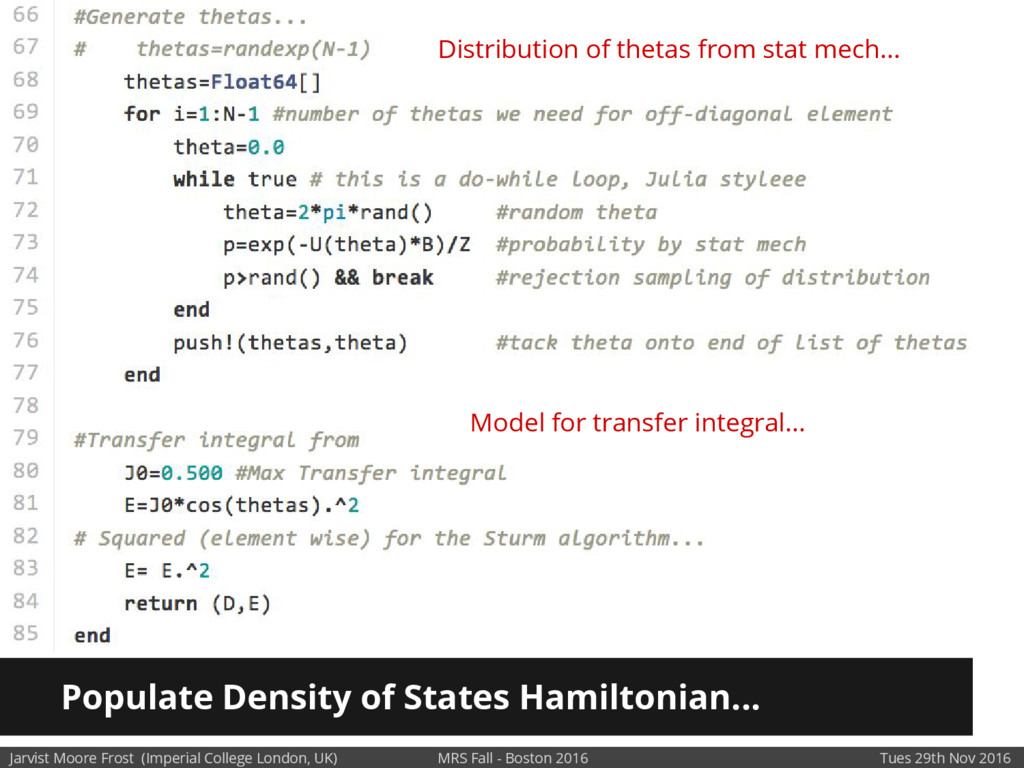
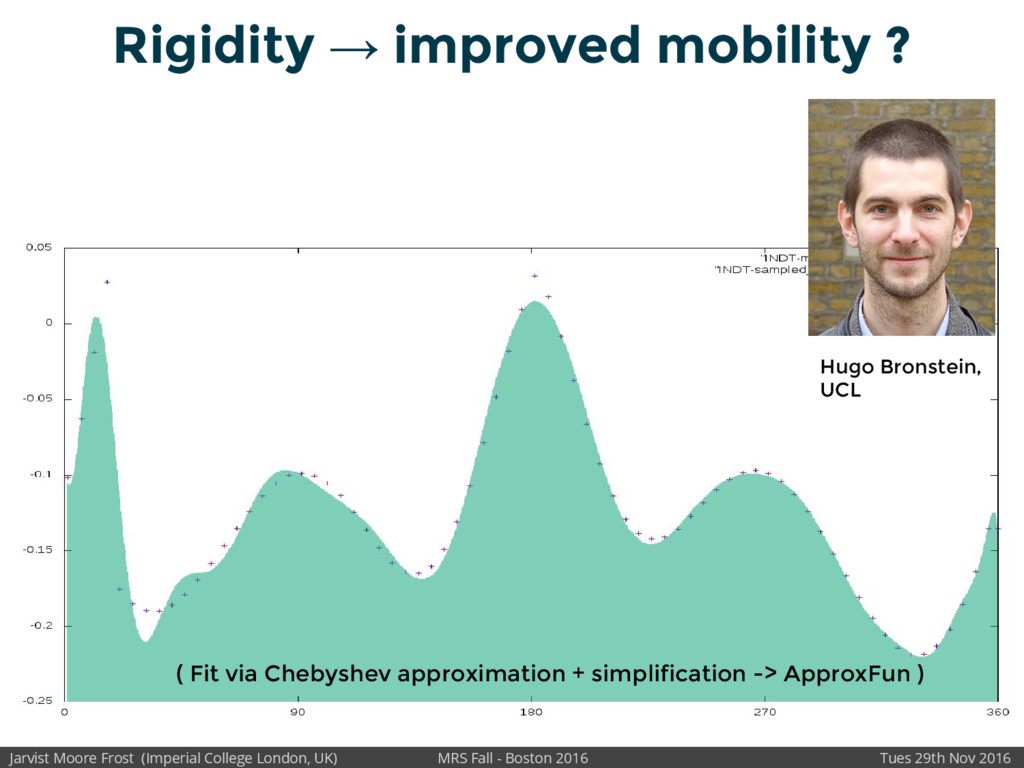
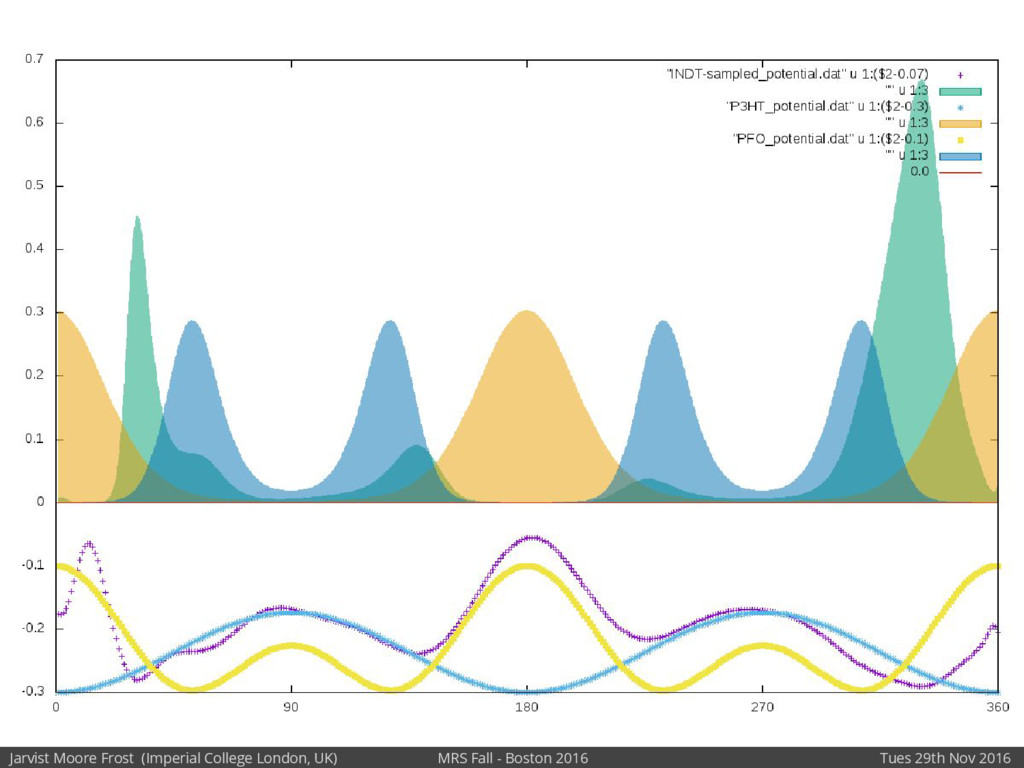
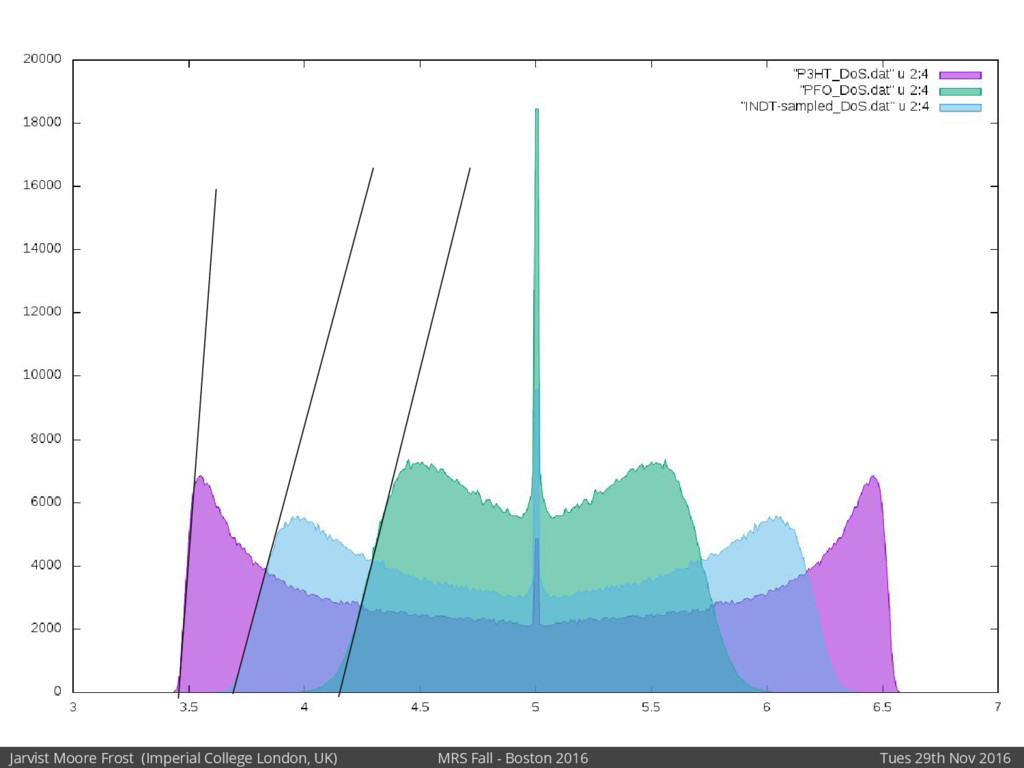
Our latest work calculates the electronic density of states for conjugated polymer chains, with the off-diagonal disorder simulated by statistical mechanics based on an ab-initio torsional potential[1,2]. The method uses the linear-scaling Sturm sequences to solve the tight binding Hamiltonian and construct the densities of states. This enables an extremely high signal to noise ratio, for minimal computational effort. This enables us to make quantitative predictions on how varying the backbone of a conjugated polymer can directly influence the resulting charge transport characteristics of both holes and electrons, and so indicate routes in chemical synthesis to materials of superior performance.
Finally we will discuss how organic materials can offer unique performance benefits over crystalline, with the use of Wavefunction Engineering to engender unique excited state properties.
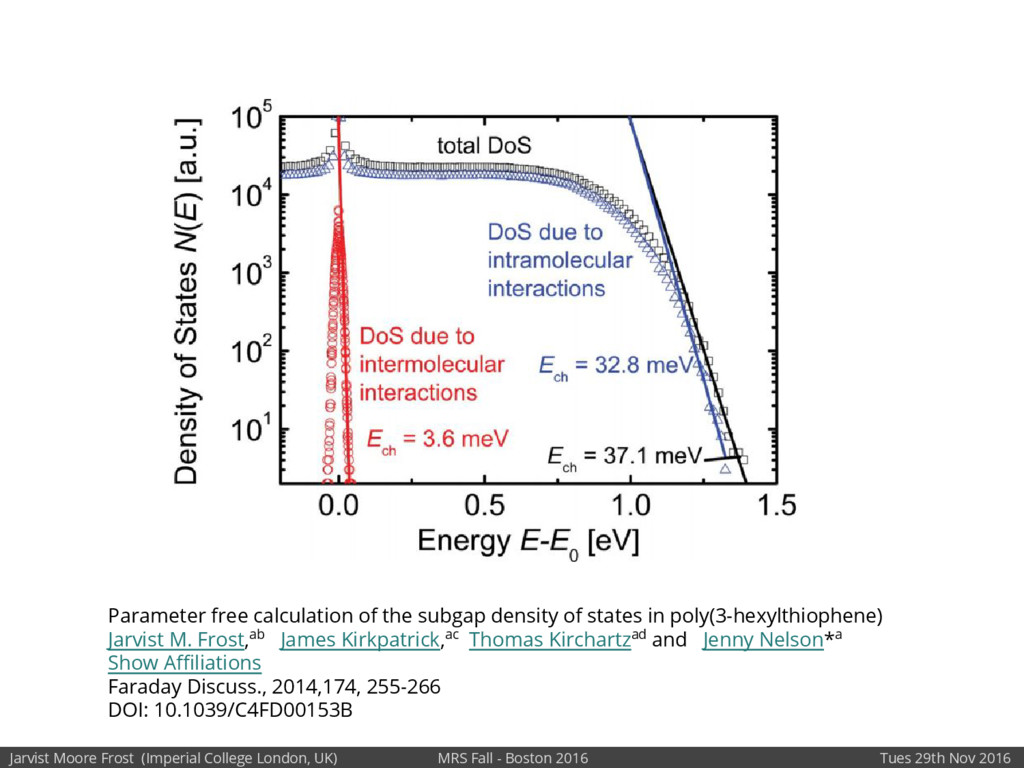
[1] Parameter free calculation of the subgap density of states in poly (3-hexylthiophene), JM Frost et al, Faraday discussions 174, 255-266
[2] https://github.com/jarvist/Teclo
EM4:
## Density of states of conjugated polymers by tight binding
### Jarvist Moore Frost, Beth Rice, Jenny Nelson
Organic electronic materials are highly spatially disordered. Resultant fluctuations in wavefunction overlap leads to band tailing in the electronic density of states. In turn this limits the charge carrier mobility. Methods to understand these relationships will enable the design of higher performance organic semiconductors.
We will discuss the multi-scale simulation methods required to solve the electronic density of states of a conjugated polymer, focusing on amorphous P3HT[1]. Here we undertook atomistic molecular dynamics, calculated transfer integrals from frozen snapshots with the molecular orbital overlap method, and then solved a tight binding model, to have an entirely ab-initio prediction of the Urbach tail of charges. Our chief results were: inter-monomer torsional disorder dominates intra-chain disorder; and that the Urbach tail was composed of extremal configurations, and so required very large calculations to converge on a value.
Our latest work calculates the electronic density of states for conjugated polymer chains, with the off-diagonal disorder simulated by statistical mechanics based on an ab-initio torsional potential[1,2]. The method uses the linear-scaling Sturm sequences to solve the tight binding Hamiltonian and construct the densities of states. This enables an extremely high signal to noise ratio, for minimal computational effort. This enables us to make quantitative predictions on how varying the backbone of a conjugated polymer can directly influence the resulting charge transport characteristics of both holes and electrons, and so indicate routes in chemical synthesis to materials of superior performance.
Finally we will discuss how organic materials can offer unique performance benefits over crystalline, with the use of Wavefunction Engineering to engender unique excited state properties.
[1] Parameter free calculation of the subgap density of states in poly (3-hexylthiophene), JM Frost et al, Faraday discussions 174, 255-266
[2] https://github.com/jarvist/Teclo
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}