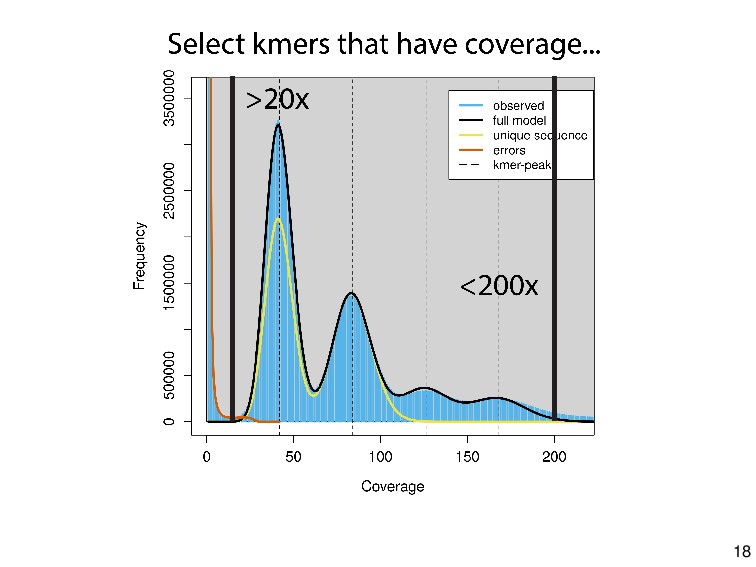
profiling from short reads. Bioinformatics 33.14 (2017): 2202-2204. Ranallo-Benavidez, T.R., Jaron, K.S. & Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11, 1432 (2020). https://doi.org/10.1038/s41467-020-14998-3 karyotype from Schwander, T., & Crespi, B. J. (2009). Multiple direct transitions from sexual reproduction to apomictic parthenogenesis in Timema stick insects. Evolution: International Journal of Organic Evolution, 63(1), 84-103. flow cytometry picture from Gutekunst, J., Andriantsoa, R., Falckenhayn, C., Hanna, K., Stein, W., Rasamy, J., & Lyko, F. (2018). Clonal genome evolution and rapid invasive spread of the marbled crayfish. Nature ecology & evolution, 2(3), 567. sequencing errors in kmers pic from https://dib-lab.github.io/zen-khmer/manipulating-histograms.html root knot phylogeny from Lunt, D. H., Kumar, S., Koutsovoulos, G., & Blaxter, M. L. (2014). The complex hybrid origins of the root knot nematodes revealed through comparative genomics. PeerJ, 2, e356. 45
{kind=link}
{kind=link}
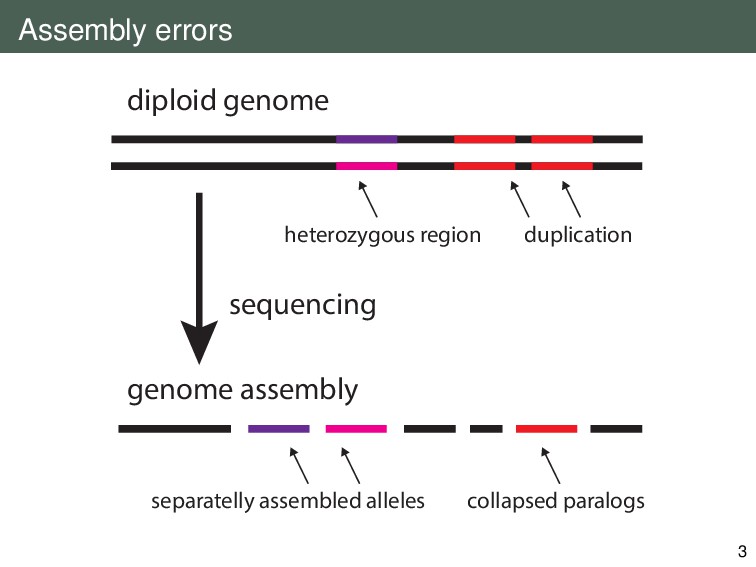{kind=link}
{kind=link}
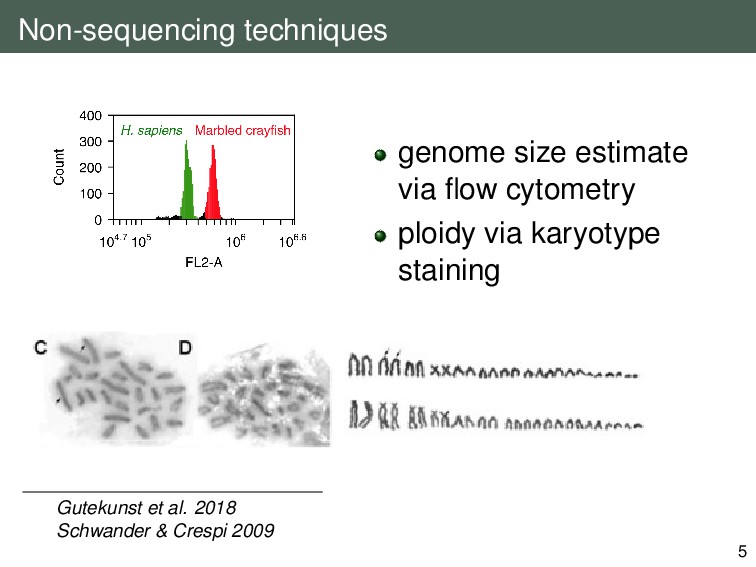{kind=link}
{kind=link}
{kind=link}
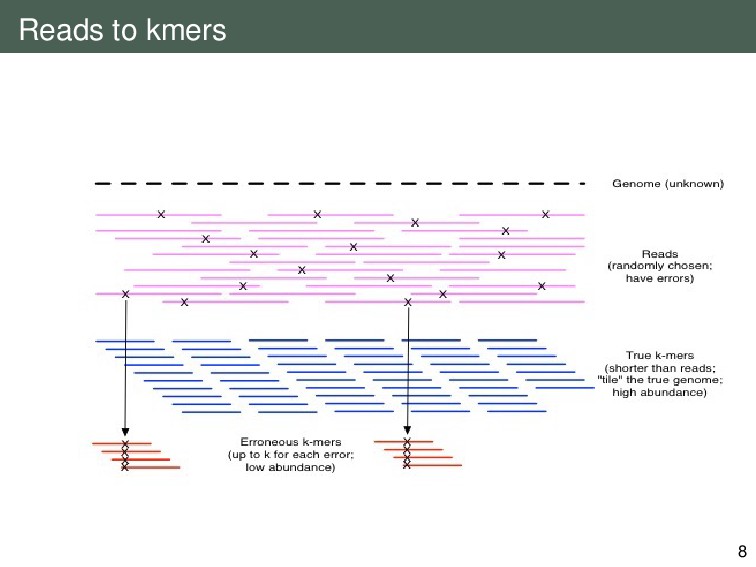{kind=link}
{kind=link}
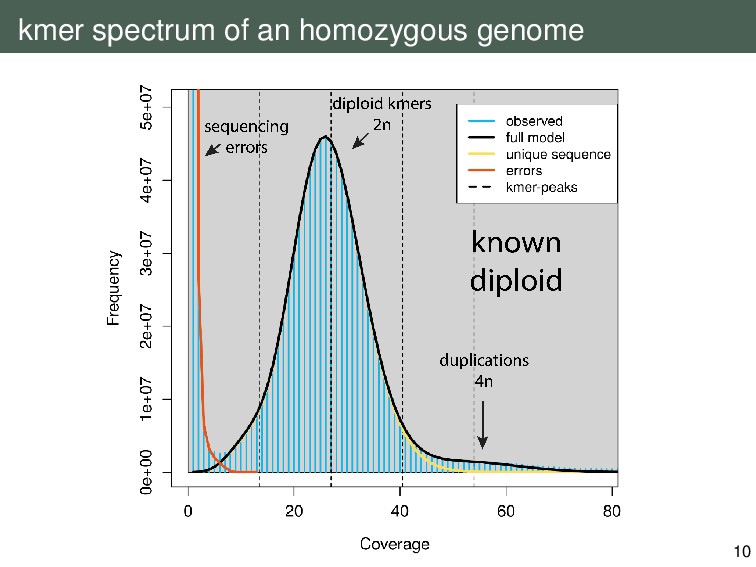{kind=link}
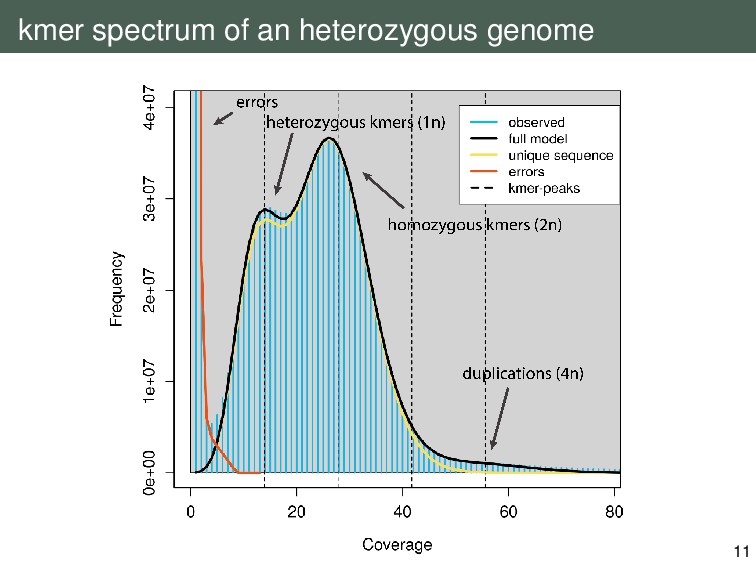{kind=link}
{kind=link}
{kind=link}
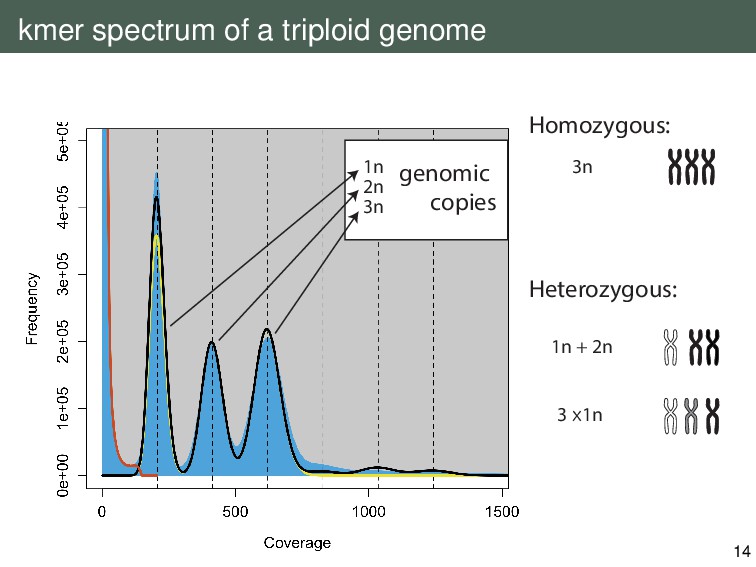{kind=link}
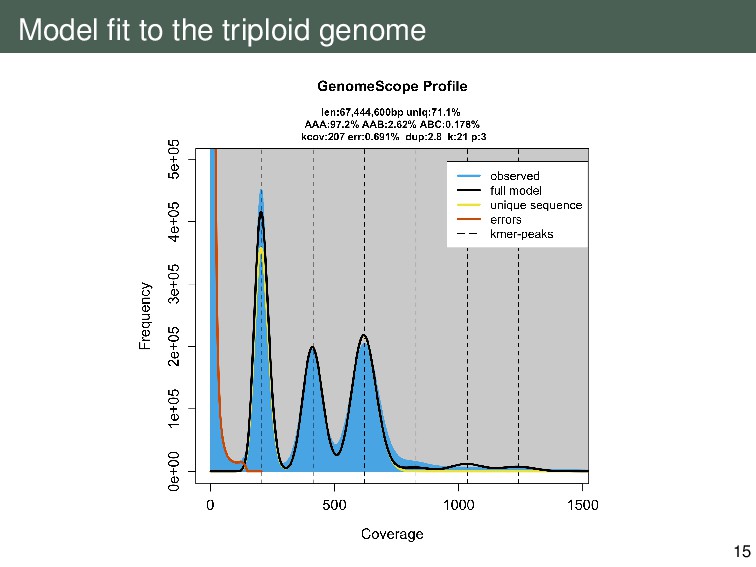{kind=link}
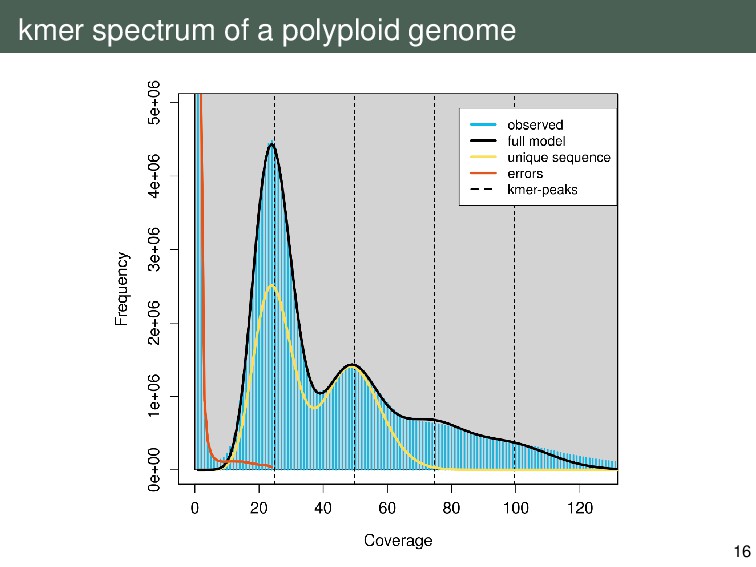{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
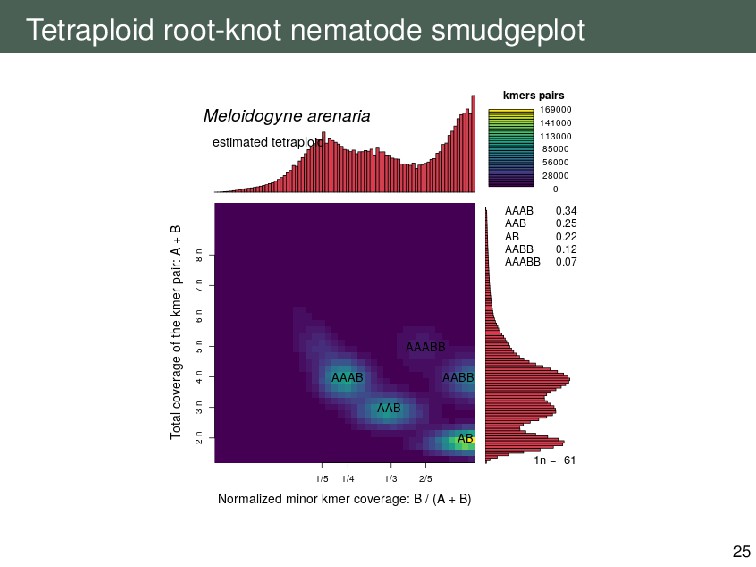{kind=link}
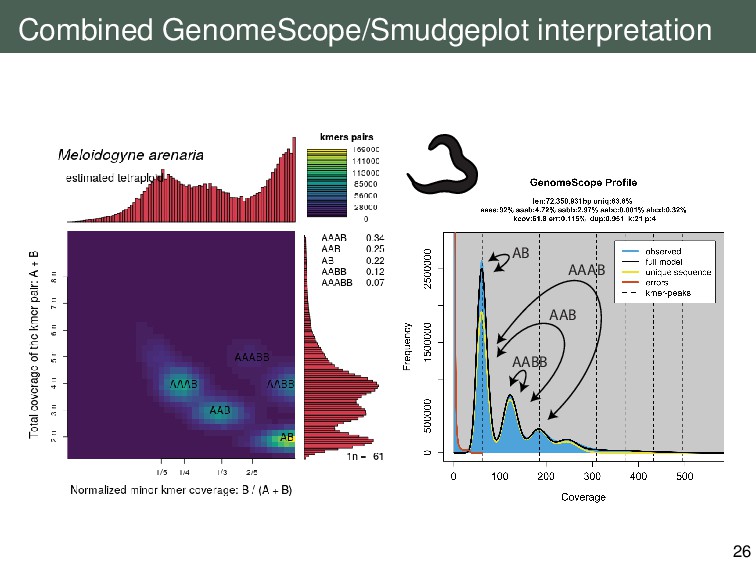{kind=link}
{kind=link}
{kind=link}
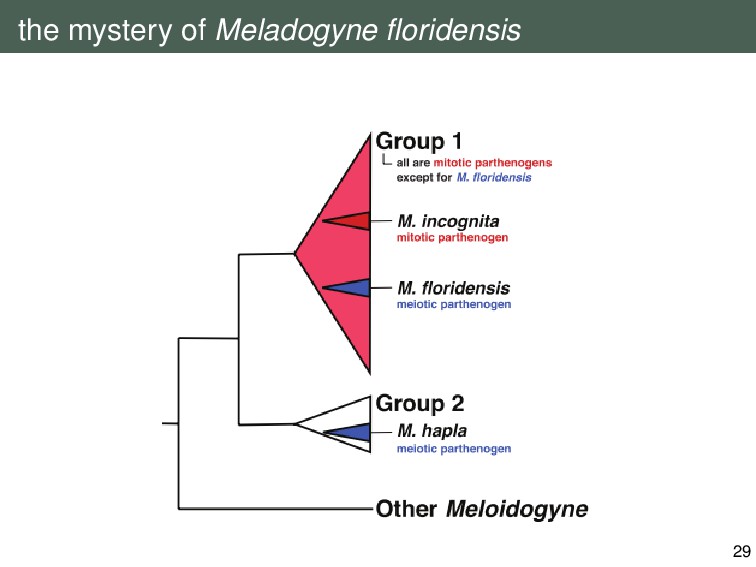{kind=link}
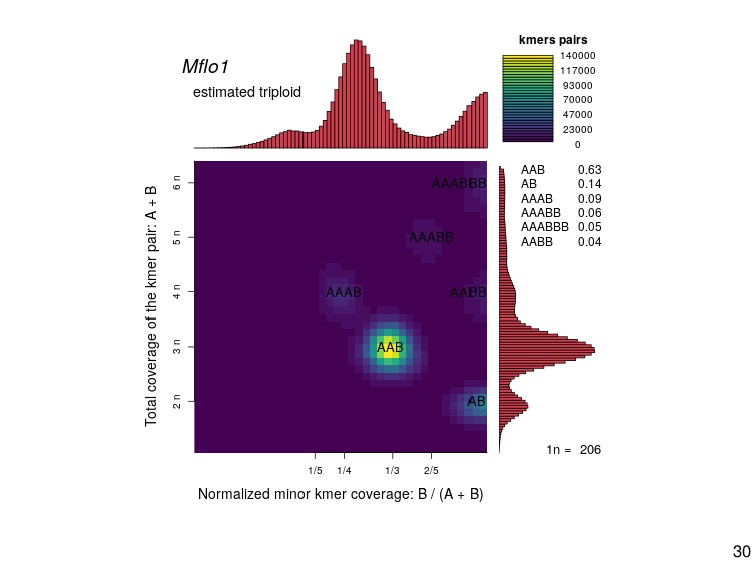{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}