Upgrade to Pro
— share decks privately, control downloads, hide ads and more …
Speaker Deck
Features
Speaker Deck
PRO
Sign in
Sign up for free
Search
Search
Operations on Genomic Intervals and Genome Arit...
Search
onouyek
December 05, 2020
Science
370
0
Share
Embed
Copy iframe code
Copy JS code
Copy link
Start on current slide
Operations on Genomic Intervals and Genome Arithmetic
【第6回】ゼロから始めるゲノム解析(R編)
onouyek
December 05, 2020
More Decks by onouyek
See All by onouyek
Finding Open Reading Frames
onouyek
0
400
Inferring mRNA from Protein: Products and Reductions of Lists
onouyek
0
380
Finding the Longest Shared Subsequence: Finding K-mers, Writing Functions, and Using Binary Search
onouyek
0
300
Find a Motif in DNA: Exploring Sequence Similarity
onouyek
0
350
Finding the Hamming Distance: Counting Point Mutations
onouyek
0
470
Creating the Fibonacci Sequence: Writing, Testing, and Benchmarking Algorithms
onouyek
1
500
Transcribing DNA into mRNA: Mutating Strings, Reading and Writing Files
onouyek
0
560
DNA methylation analysis using bisulfite sequencing data
onouyek
0
920
Exploratory Data Analysis with Unsupervised Machine Learning
onouyek
0
320
Other Decks in Science
See All in Science
人生を変えた一冊「独学大全」のはなし / Self-study ENCYCLOPEDIA: The Book Which Change My Life #独学大全 #EM推し本
expajp
0
170
機械学習 - pandas入門
trycycle
PRO
0
650
見上公一.pdf
genomethica
0
160
HajimetenoLT vol.17
hashimoto_kei
1
240
[NLP2026 参加報告会] AI for Science まとめ / NLP2026
lychee1223
0
1.9k
AkarengaLT vol.41
hashimoto_kei
1
150
Non-Gaussian, nonlinear causal discovery with hidden variables and application
sshimizu2006
0
150
サンプル対応のない複数遺伝子発現プロファイルに対するテンソル分解型統合解析の要約
tagtag
PRO
0
210
俺たちは本当に分かり合えるのか? ~ PdMとスクラムチームの “ずれ” を科学する
bonotake
2
2.5k
検索と推論タスクに関する論文の紹介
ynakano
1
250
あなたに水耕栽培を愛していないとは言わせない
mutsumix
1
360
(2025) Balade en cyclotomie
mansuy
0
640
Featured
See All Featured
Measuring Dark Social's Impact On Conversion and Attribution
stephenakadiri
2
230
Designing for Performance
lara
611
70k
The Pragmatic Product Professional
lauravandoore
37
7.4k
Amusing Abliteration
ianozsvald
1
220
16th Malabo Montpellier Forum Presentation
akademiya2063
PRO
0
170
Faster Mobile Websites
deanohume
310
32k
Scaling GitHub
holman
464
140k
Build your cross-platform service in a week with App Engine
jlugia
234
18k
Jamie Indigo - Trashchat’s Guide to Black Boxes: Technical SEO Tactics for LLMs
techseoconnect
PRO
0
260
Visual Storytelling: How to be a Superhuman Communicator
reverentgeek
2
590
Rebuilding a faster, lazier Slack
samanthasiow
85
9.6k
Reality Check: Gamification 10 Years Later
codingconduct
0
2.2k
Transcript
Operations on Genomic Intervals and Genome Arithmetic 第6回ゼロから始めるゲノム解析(後半) onouyek@2020年12月4日
データセットの準備 library(GenomicRanges) devtools::install_github("compgenomr/compGenomRData") # CpGアイランドのデータパスを取得 filePath <- system.file("extdata", "cpgi.hg19.chr21.bed", package="compGenomRData")
# CpGアイランドのデータを読み込み cpgi.df <- read.table(filePath, header=FALSE, stringsAsFactors=FALSE) # "_"を含む染色体名を除外 cpgi.df <- cpgi.df [grep("_",cpgi.df[,1],invert=TRUE),] cpgi.gr <- GRanges(seqnames=cpgi.df[,1], ranges=IRanges(start=cpgi.df[,2], end=cpgi.df[,3]))
詳細情報を含む ゲノム間隔 Genomic intervals with more information
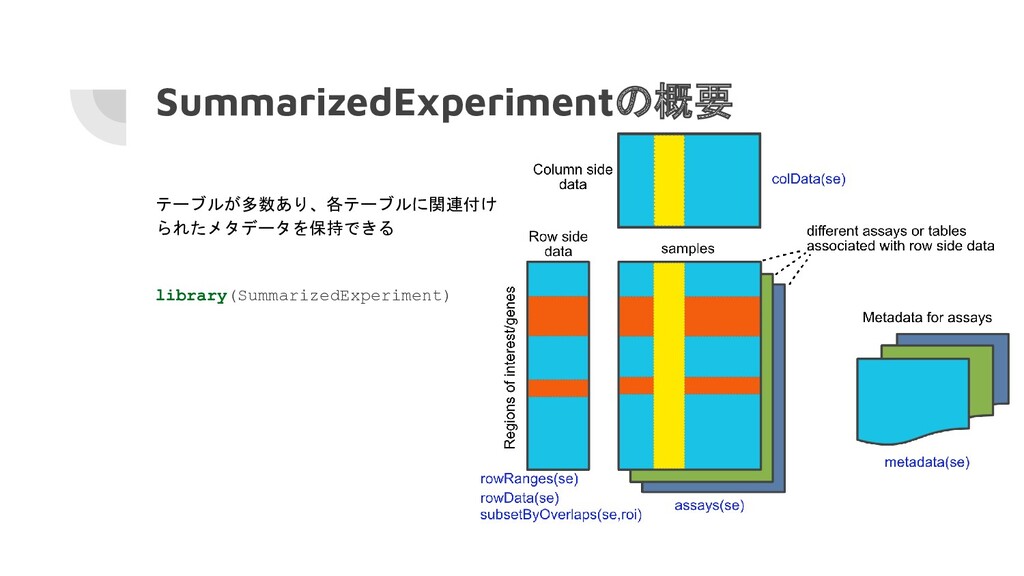
SummarizedExperimentの概要 テーブルが多数あり、各テーブルに関連付け られたメタデータを保持できる library(SummarizedExperiment)
SummarizedExperimentオブジェクトの作成 # RNA-seq のリードカウントテーブルを想定 nrows <- 200 ncols <- 6
counts <- matrix(runif(nrows * ncols, 1, 1e4), nrows) # 遺伝子の位置オブジェクトの作成 rowRanges <- GRanges(rep(c("chr1", "chr2"), c(50, 150)), IRanges(floor(runif(200, 1e5, 1e6)), width=100), strand=sample(c("+", "-"), 200, TRUE), feature_id=paste0("gene", 1:200)) # 列名のためのデータフレームを作成 colData <- DataFrame(timepoint=1:6, row.names=LETTERS[1:6])
SummarizedExperimentオブジェクトの作成 # SummarizedExperimentオブジェクトの作成 se <- SummarizedExperiment(assays=list(counts=counts), rowRanges=rowRanges, colData=colData) se ##
class: RangedSummarizedExperiment ## dim: 200 6 ## metadata(0): ## assays(1): counts ## rownames: NULL ## rowData names(1): feature_id ## colnames(6): A B ... E F ## colData names(1): timepoint
SummarizedExperimentオブジェクトの抽出 # 列の抽出 colData(se) ## DataFrame with 6 rows and
1 column ## timepoint ## <integer> ## A 1 ## B 2 ## C 3 ## D 4 ## E 5 ## F 6 # 行の抽出 rowData(se) ## DataFrame with 200 rows and 1 column ## feature_id ## <character> ## 1 gene1 ## 2 gene2 ## 3 gene3 ## 4 gene4 ## 5 gene5 ## ... ... ## 196 gene196 ## 197 gene197 ## 198 gene198 ## 199 gene199 ## 200 gene200
SummarizedExperimentオブジェクトの抽出 # 1つ以上のテーブルリストを抽出 assays(se) ## List of length 1 ##
names(1): counts # $を用いて"counts"テーブルを抽出 assays(se)$counts # []を用いて最初のテーブルを抽出 assays(se)[[1]]
SummarizedExperimentオブジェクトのサブ セット化 # 最初の5つの転写物と最初の3つのサンプルを サブセット化 se[1:5, 1:3] ## class: RangedSummarizedExperiment
## dim: 5 3 ## metadata(0): ## assays(1): counts ## rownames: NULL ## rowData names(1): feature_id ## colnames(3): A B C ## colData names(1): timepoint # chr1:100,000-1,100,000の行でサブセット化 roi <- GRanges(seqnames="chr1", ranges=100000:1100000) subsetByOverlaps(se, roi) ## class: RangedSummarizedExperiment ## dim: 50 6 ## metadata(0): ## assays(1): counts ## rownames: NULL ## rowData names(1): feature_id ## colnames(6): A B ... E F ## colData names(1): timepoint
ゲノム間隔の 視覚化と要約 Visualizing and summarizing genomic intervals
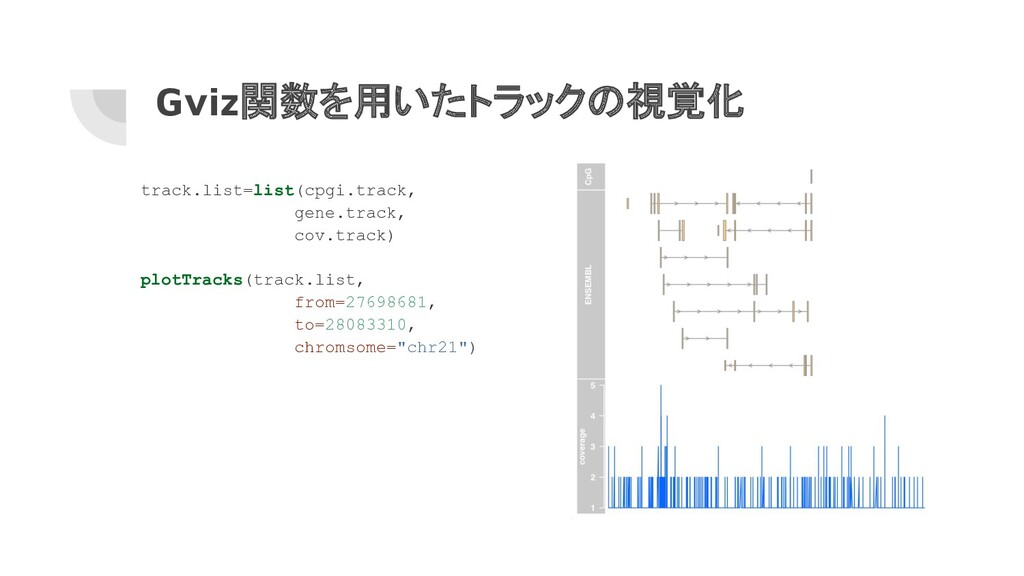
Gviz関数を用いたトラックの設定 library(Gviz) # CpGアイランドのトラックを設定 cpgi.track <- AnnotationTrack(cpgi.gr, name="CpG") # 遺伝子のトラックを設定
# library(httr) # httr::set_config(config(ssl_verifypeer=FALSE)) gene.track <- BiomartGeneRegionTrack(genome="hg19", chromosome="chr21", start=27698681, end=28083310, name="ENSEMBL") # ChIP-seqのカバレッジトラックを設定 chipseqFile <- system.file("extdata", "wgEncodeHaibTfbsA549.chr21.bw", package="compGenomRData") cov.track <- DataTrack(chipseqFile, type="l", name="coverage")
Gviz関数を用いたトラックの視覚化 track.list=list(cpgi.track, gene.track, cov.track) plotTracks(track.list, from=27698681, to=28083310, chromsome="chr21")
複数の遺伝子座のゲノム間隔の要約 library(genomation) # 染色体番号20の転写開始部位のデータを取得 transcriptFile <- system.file("extdata", "refseq.hg19.chr20.bed", package="compGenomRData") feat
<- readTranscriptFeatures(transcriptFile, remove.unusual=TRUE, up.flank=500, down.flank=500) # プロモーターの情報を取得 prom <- feat$promoters # 転写開始部位周辺のH3K4me3の情報を取得 H3K4me3File <- system.file("extdata", "H1.ESC.H3K4me3.chr20.bw", package="compGenomRData") sm <- ScoreMatrix(H3K4me3File, prom, type="bigWig", strand.aware=TRUE)
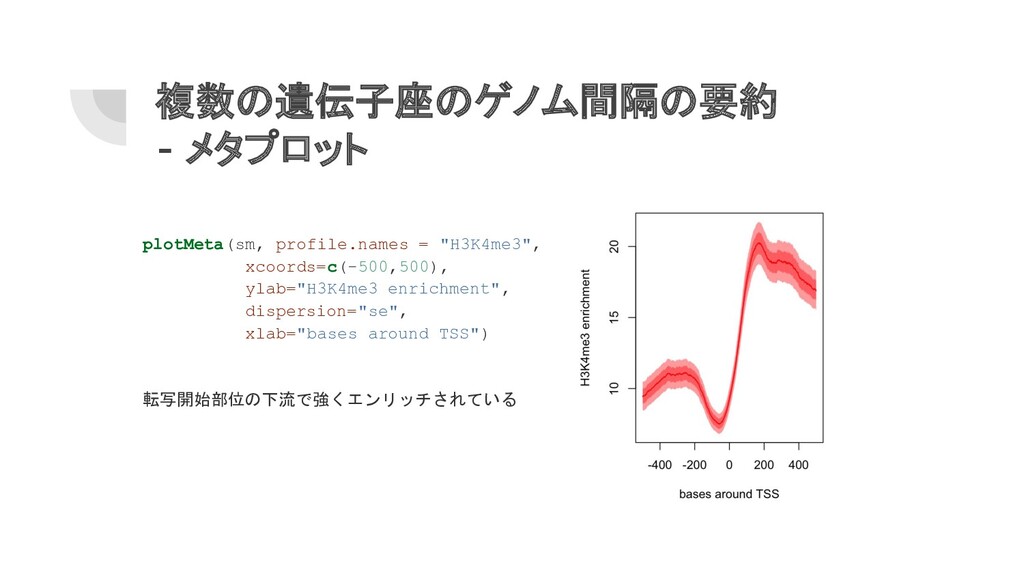
複数の遺伝子座のゲノム間隔の要約 - メタプロット plotMeta(sm, profile.names = "H3K4me3", xcoords=c(-500,500), ylab="H3K4me3 enrichment",
dispersion="se", xlab="bases around TSS") 転写開始部位の下流で強くエンリッチされている
複数の遺伝子座のゲノム間隔の要約 - ヒートマップ heatMatrix(sm, order=TRUE, xcoords=c(-500,500), xlab="bases around TSS") 各行が転写開始部位周囲の領域
エンリッチメントの強度で色分けされている
複数の遺伝子座のゲノム間隔の要約 - メタプロットの組み合わせ # DNAseのデータを組み合わせる DNAseFile <- system.file("extdata", "H1.ESC.dnase.chr20.bw", package="compGenomRData")
sml <- ScoreMatrixList(c(H3K4me3=H3K4me3File, DNAse=DNAseFile), prom, type="bigWig", strand.aware=TRUE) plotMeta(sml)
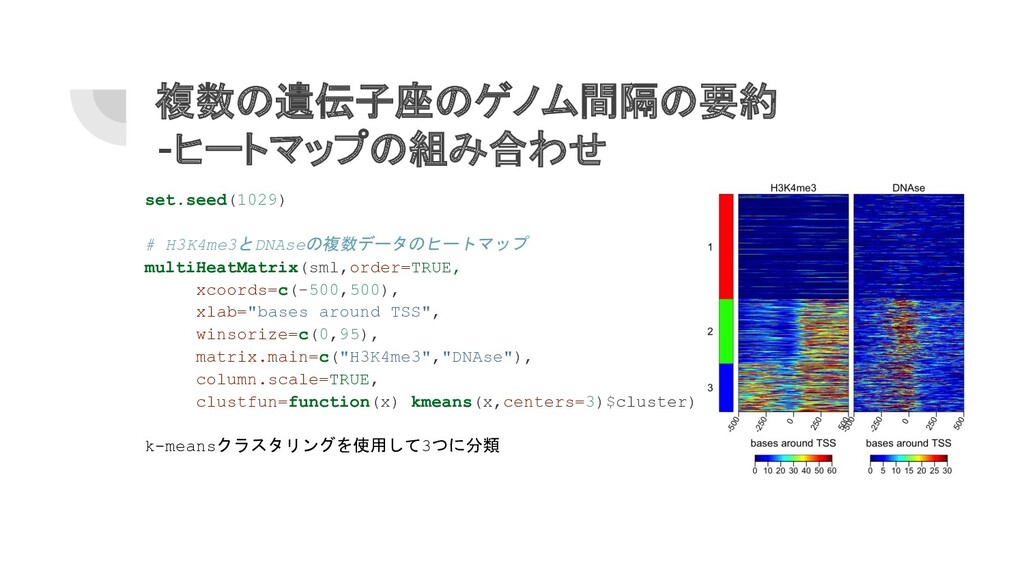
複数の遺伝子座のゲノム間隔の要約 -ヒートマップの組み合わせ set.seed(1029) # H3K4me3とDNAseの複数データのヒートマップ multiHeatMatrix(sml,order=TRUE, xcoords=c(-500,500), xlab="bases around TSS",
winsorize=c(0,95), matrix.main=c("H3K4me3","DNAse"), column.scale=TRUE, clustfun=function(x) kmeans(x,centers=3)$cluster) k-meansクラスタリングを使用して3つに分類
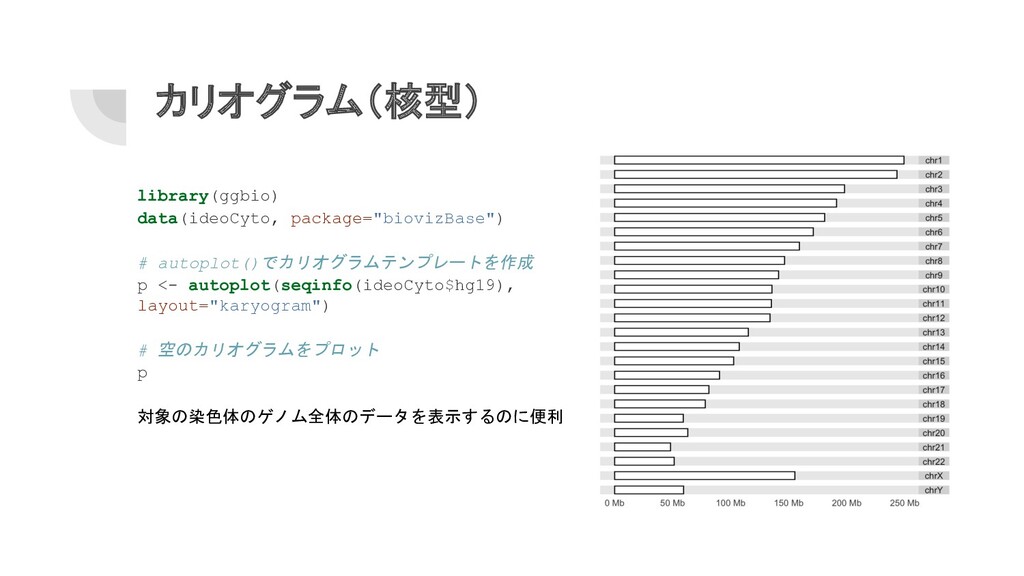
カリオグラム(核型) library(ggbio) data(ideoCyto, package="biovizBase") # autoplot()でカリオグラムテンプレートを作成 p <- autoplot(seqinfo(ideoCyto$hg19), layout="karyogram")
# 空のカリオグラムをプロット p 対象の染色体のゲノム全体のデータを表示するのに便利
カリオグラム + CpGアイランド CpGiFile <- system.file("extdata", "CpGi.hg19.table.txt", package="compGenomRData") # CpGアイランドの情報を読み込み
cpgi.gr <- genomation::readGeneric(CpGiFile, chr=1, start=2, end=3, header=TRUE, keep.all.metadata=TRUE, remove.unusual=TRUE) # layout_karyogram()でCpGアイランドをプロット p + layout_karyogram(cpgi.gr)
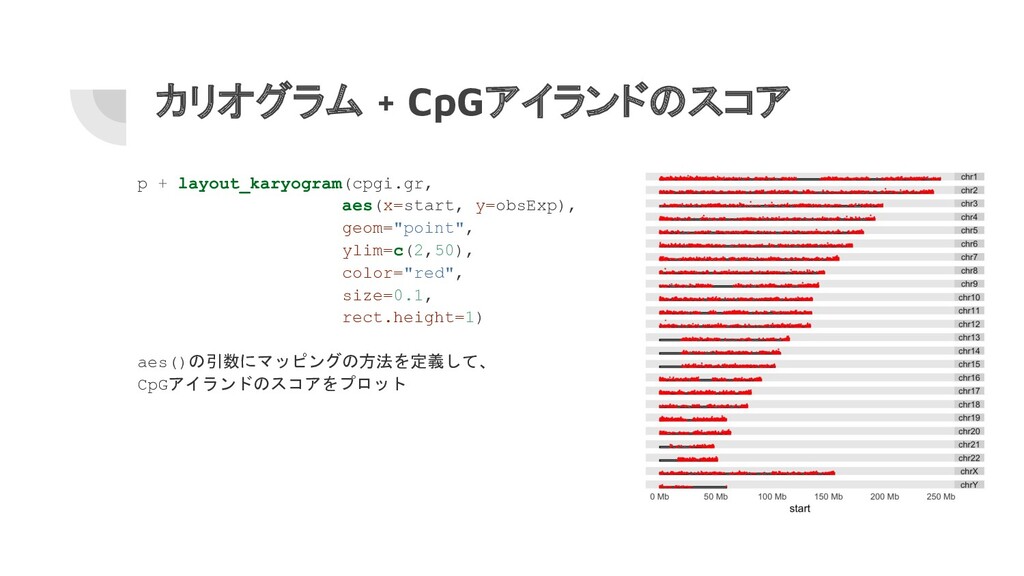
カリオグラム + CpGアイランドのスコア p + layout_karyogram(cpgi.gr, aes(x=start, y=obsExp), geom="point", ylim=c(2,50),
color="red", size=0.1, rect.height=1) aes()の引数にマッピングの方法を定義して、 CpGアイランドのスコアをプロット
Circosプロット # サークルの中に染色体を設定 p <- ggplot() + layout_circle(ideoCyto$hg19, geom =
"ideo", fill = "white", colour = "white", cytoband = TRUE, radius = 39, trackWidth = 2) # CpGアイランドのスコアをドットに設定 p <- p + layout_circle(cpgi.gr, geom = "point", grid=TRUE, size = 0.01, aes(y = obsExp),color="red", radius = 42, trackWidth = 10) # 染色体名を設定 p <- p + layout_circle(as(seqinfo(ideoCyto$hg19),"GRanges"), geom = "text", aes(label = seqnames), vjust = 0, radius = 55, trackWidth = 7, size=3) # プロットを表示 p
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![SummarizedExperimentオブジェクトのサブ セット化 # 最初の5つの転写物と最初の3つのサンプルを サブセット化 se[1:5, 1:3] ## class: RangedSummarizedExperiment](https://files.speakerdeck.com/presentations/86df8188afa54e09a37e809799d2b716/slide_8.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}