Upgrade to Pro
— share decks privately, control downloads, hide ads and more …
Speaker Deck
Features
Speaker Deck
PRO
Sign in
Sign up for free
Search
Search
Exploratory Data Analysis with Unsupervised Mac...
Search
onouyek
October 23, 2020
Technology
320
0
Share
Embed
Copy iframe code
Copy JS code
Copy link
Start on current slide
Exploratory Data Analysis with Unsupervised Machine Learning
【第4回】ゼロから始めるゲノム解析(R編)
onouyek
October 23, 2020
More Decks by onouyek
See All by onouyek
Finding Open Reading Frames
onouyek
0
400
Inferring mRNA from Protein: Products and Reductions of Lists
onouyek
0
380
Finding the Longest Shared Subsequence: Finding K-mers, Writing Functions, and Using Binary Search
onouyek
0
300
Find a Motif in DNA: Exploring Sequence Similarity
onouyek
0
350
Finding the Hamming Distance: Counting Point Mutations
onouyek
0
470
Creating the Fibonacci Sequence: Writing, Testing, and Benchmarking Algorithms
onouyek
1
500
Transcribing DNA into mRNA: Mutating Strings, Reading and Writing Files
onouyek
0
560
DNA methylation analysis using bisulfite sequencing data
onouyek
0
920
Operations on Genomic Intervals and Genome Arithmetic
onouyek
0
370
Other Decks in Technology
See All in Technology
SRE歴2ヶ月でも開発6年の知見を活かして、チームで止まっていた環境改善を前に進めた話
a_ono
1
220
Claude Code 珍プレー好プレー
shinyasaita
0
280
Text-to-SQLをAgentCoreで実現し、生成されるSQLの精度を定量的に評価する
yakumo
2
660
AWS Blocks を触ってみた/first-tach-aws-blocks
fossamagna
2
150
Mastraエージェント、どのクラウドにデプロイする?
minorun365
PRO
2
170
AI時代における最適なQA組織の作り方
ymty
3
460
AI時代のエンジニアキャリアについて今一度考える
sakamoto_582
2
1.3k
AIで政治は変わるのか? — 中高生と考えたAI時代の民主主義(東海高校サタデープログラム)
eitarosuda
0
400
最適な自走を最小限の支援で — M&Aで拡大する組織で少人数SREが挑んだ1年 / SRE NEXT 2026
genda
0
280
アカウントが増えてからでは遅い? ~ マルチアカウント統制の勘所 ~
kenichinakamura
0
190
Claude Codeとハーネスについて考えてみる
oikon48
18
8.8k
AWS Summit の片隅で、体育座りしながらコミュニティがにぎわう理由を考えた
k_adachi_01
2
360
Featured
See All Featured
エンジニアに許された特別な時間の終わり
watany
107
250k
How to Build an AI Search Optimization Roadmap - Criteria and Steps to Take #SEOIRL
aleyda
1
2.1k
Understanding Cognitive Biases in Performance Measurement
bluesmoon
32
3k
A Guide to Academic Writing Using Generative AI - A Workshop
ks91
PRO
1
340
The agentic SEO stack - context over prompts
schlessera
0
840
Tips & Tricks on How to Get Your First Job In Tech
honzajavorek
1
550
Embracing the Ebb and Flow
colly
88
5.1k
How to Align SEO within the Product Triangle To Get Buy-In & Support - #RIMC
aleyda
2
1.6k
Crafting Experiences
bethany
1
210
Practical Tips for Bootstrapping Information Extraction Pipelines
honnibal
25
2k
Google's AI Overviews - The New Search
badams
0
1.1k
The Organizational Zoo: Understanding Human Behavior Agility Through Metaphoric Constructive Conversations (based on the works of Arthur Shelley, Ph.D)
kimpetersen
PRO
0
380
Transcript
Exploratory Data Analysis with Unsupervised Machine Learning 第4回ゼロから始めるゲノム解析(R編) onouyek@2020年10月23日
クラスタリング
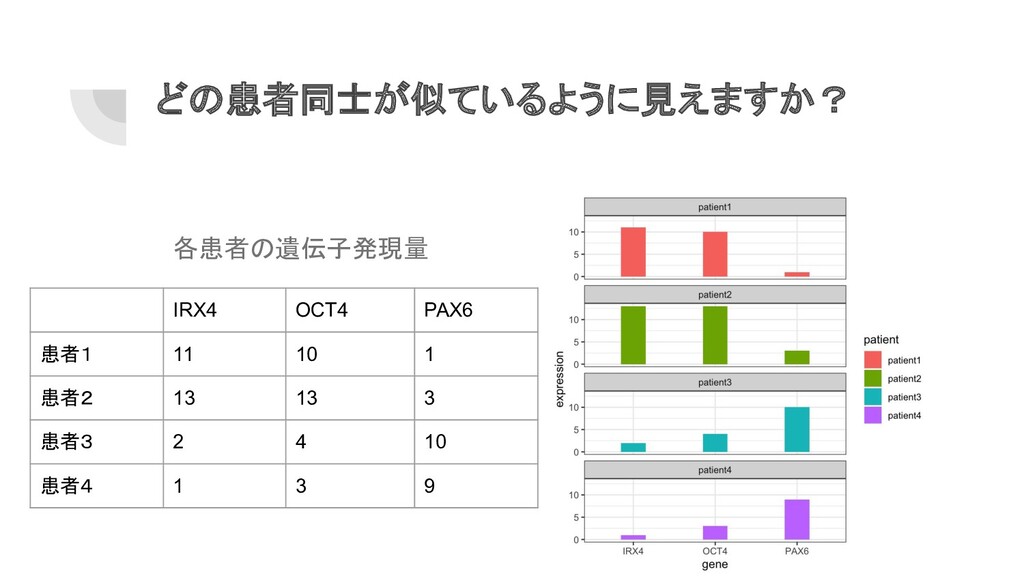
どの患者同士が似ているように見えますか? 各患者の遺伝子発現量 IRX4 OCT4 PAX6 患者1 11 10 1 患者2
13 13 3 患者3 2 4 10 患者4 1 3 9
距離概念 マンハッタン距離(L1ノルム) 各遺伝子発現量の患者Aと患者Bの差の絶対 値の合計 # マンハッタン距離 dist(df,method="manhattan") ## patient1 patient2
patient3 ## patient2 7 ## patient3 24 27 ## patient4 25 28 3
距離概念 ユーグリッド距離(L2ノルム) 各遺伝子発現量の患者Aと患者Bの差の2乗の 合計の平方根 2乗操作により、非常に異なる値は距離への寄 与が大きくなる。 マンハッタン距離と比較して外れ値の影響を受 けやすい。 # ユーグリッド距離
dist(df,method="euclidean") ## patient1 patient2 patient3 ## patient2 4.123106 ## patient3 14.071247 15.842980 ## patient4 14.594520 16.733201 1.732051
距離概念 相関距離 1 − ピアソンの相関係数 発現が似ている場合は相関が高いので、相 関距離は小さくなる 発現パターンが異なる場合は相関距離は大 きくなる #
相関距離 as.dist(1-cor(t(df))) ## patient1 patient2 patient3 ## patient2 0.004129405 ## patient3 1.988522468 1.970725343 ## patient4 1.988522468 1.970725343 0.000000000
スケーリング ある遺伝子の発現量が他の遺伝子の発現量よ りもはるかに高い場合、スケーリング(正規化) をする必要がある。 ただし、スケーリングを適用するかどうかはデー タと何を目的とするかによる。 scale(df) ## IRX4 OCT4
PAX6 ## patient1 0.6932522 0.5212860 -1.0733721 ## patient2 1.0194886 1.1468293 -0.6214260 ## patient3 -0.7748113 -0.7298004 0.9603856 ## patient4 -0.9379295 -0.9383149 0.7344125 ## attr(,"scaled:center") ## IRX4 OCT4 PAX6 ## 6.75 7.50 5.75 ## attr(,"scaled:scale") ## IRX4 OCT4 PAX6 ## 6.130525 4.795832 4.425306
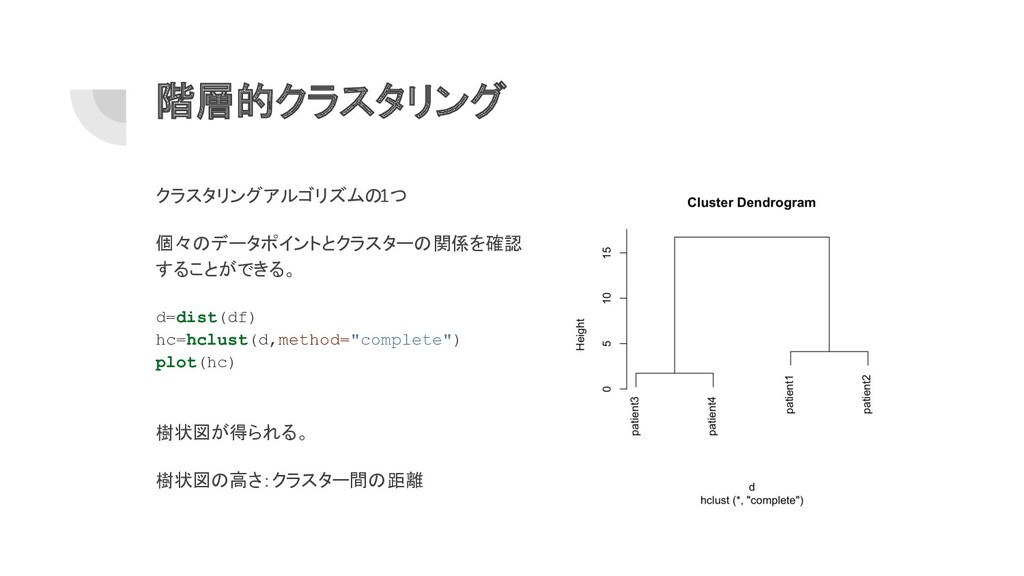
階層的クラスタリング クラスタリングアルゴリズムの 1つ 個々のデータポイントとクラスターの関係を確認 することができる。 d=dist(df) hc=hclust(d,method="complete") plot(hc) 樹状図が得られる。 樹状図の高さ:クラスター間の距離
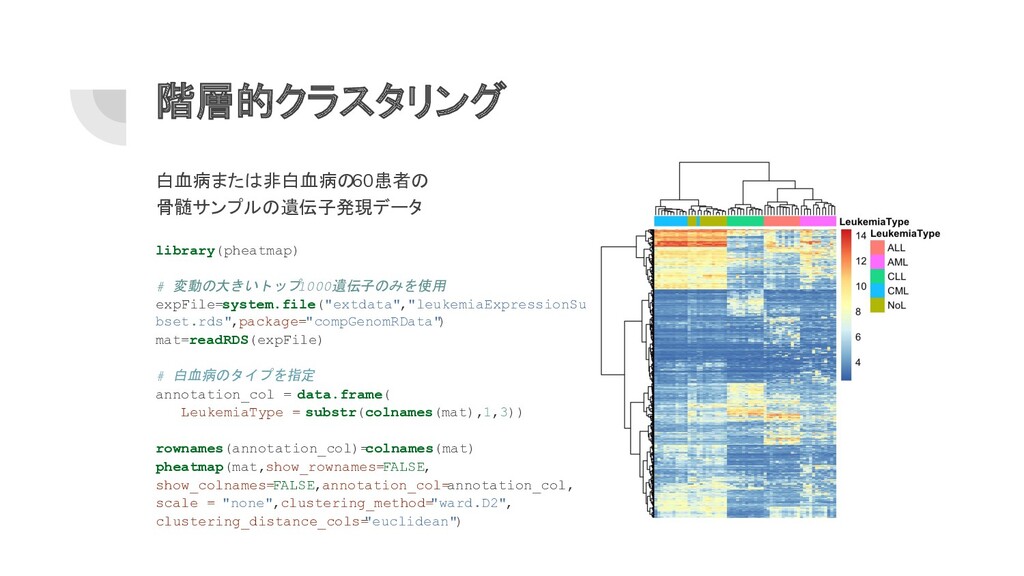
階層的クラスタリング 白血病または非白血病の 60患者の 骨髄サンプルの遺伝子発現データ library(pheatmap) # 変動の大きいトップ 1000遺伝子のみを使用 expFile=system.file("extdata","leukemiaExpressionSu bset.rds",package="compGenomRData"
) mat=readRDS(expFile) # 白血病のタイプを指定 annotation_col = data.frame( LeukemiaType = substr(colnames(mat),1,3)) rownames(annotation_col)= colnames(mat) pheatmap(mat,show_rownames= FALSE, show_colnames= FALSE,annotation_col= annotation_col, scale = "none",clustering_method= "ward.D2", clustering_distance_cols= "euclidean")
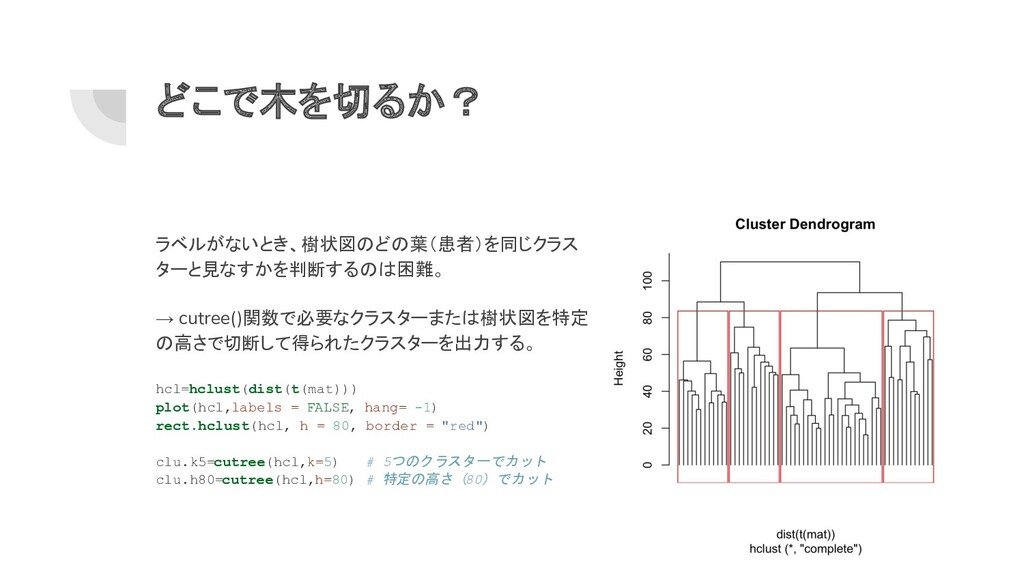
どこで木を切るか? ラベルがないとき、樹状図のどの葉(患者)を同じクラス ターと見なすかを判断するのは困難。 → cutree()関数で必要なクラスターまたは樹状図を特定 の高さで切断して得られたクラスターを出力する。 hcl=hclust(dist(t(mat))) plot(hcl,labels = FALSE,
hang= -1) rect.hclust(hcl, h = 80, border = "red") clu.k5=cutree(hcl,k=5) # 5つのクラスターでカット clu.h80=cutree(hcl,h=80) # 特定の高さ(80)でカット
k-meansクラスタリング クラスタリングアルゴリズムの 1つ データを事前に決定された k個のクラスターに分割する方 法 「パーティション化」メソッドと呼ばれる。 1. 初期化段階では実際の患者の遺伝子発現分布の境 界内でランダムに選択される。
2. 各患者は最も近い重心に割り当てられる。 3. 重心がクラスター内の遺伝子の値の平均に設定さ れる。 4. クラスターの重心までの距離の2乗の合計が最小 になるまで繰り返される。 set.seed(101) # kmeans()で患者間の距離を計算 kclu=kmeans(t(mat),centers=5) # 各クラスターのデータポイント数を確認 table(kclu$cluster) ## ## 1 2 3 4 5 ## 12 14 11 12 11
k-medoidsクラスタリング 基本的な手順はk-meansクラスタリングと同じ 選択された重心は、症例患者の実際のデータポイント 各反復で最適化しようとしているメトリックは重心までのマン ハッタン距離に基づいている。(k-meansではユーグリッド距 離) 各クラスター内の各白血病タイプの割合を確認 kmclu=cluster::pam(t(mat),k=5) type2kmclu =
data.frame( LeukemiaType=substr(colnames(mat),1,3), cluster=kmclu$cluster) table(type2kmclu) ## cluster ## LeukemiaType 1 2 3 4 5 ## ALL 12 0 0 0 0 ## AML 0 10 1 1 0 ## CLL 0 0 0 0 12 ## CML 0 0 0 12 0 ## NoL 0 0 12 0 0
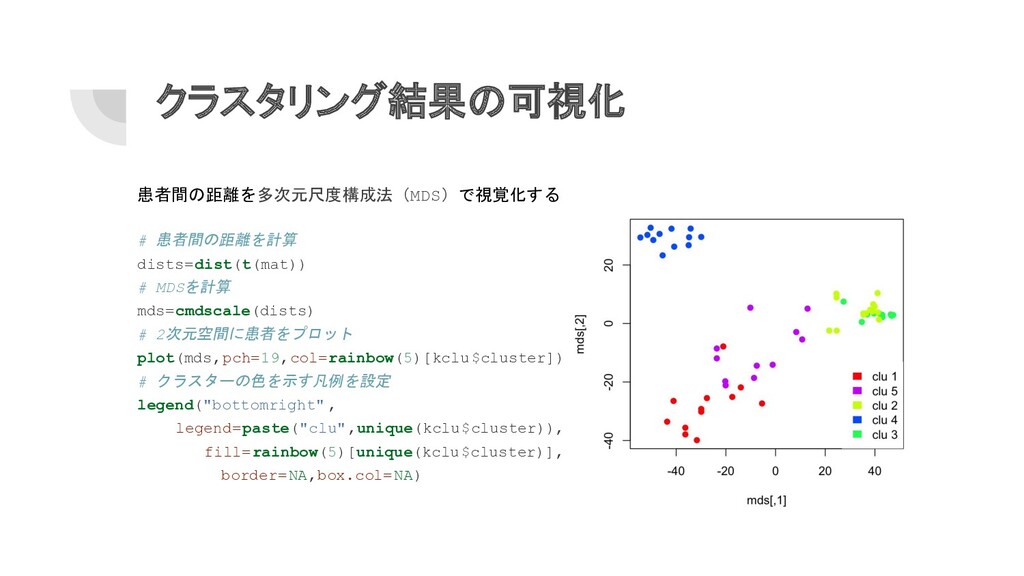
クラスタリング結果の可視化 患者間の距離を多次元尺度構成法(MDS)で視覚化する # 患者間の距離を計算 dists=dist(t(mat)) # MDSを計算 mds=cmdscale(dists) # 2次元空間に患者をプロット
plot(mds,pch=19,col=rainbow(5)[kclu$cluster]) # クラスターの色を示す凡例を設定 legend("bottomright" , legend=paste("clu",unique(kclu$cluster)), fill=rainbow(5)[unique(kclu$cluster)], border=NA,box.col=NA)
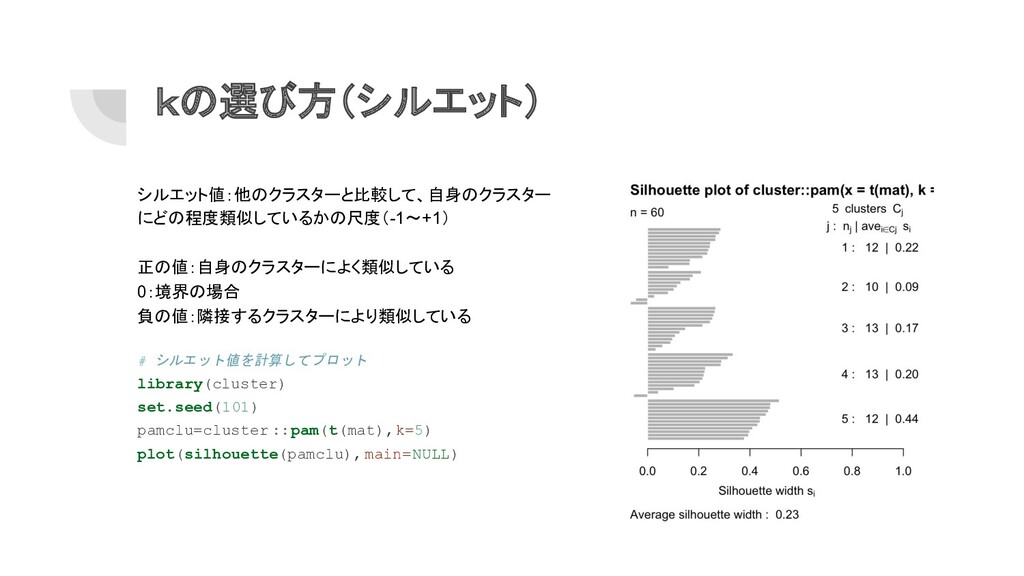
kの選び方(シルエット) シルエット値:他のクラスターと比較して、自身のクラスター にどの程度類似しているかの尺度(-1〜+1) 正の値:自身のクラスターによく類似している 0:境界の場合 負の値:隣接するクラスターにより類似している # シルエット値を計算してプロット library(cluster) set.seed(101)
pamclu=cluster ::pam(t(mat),k=5) plot(silhouette(pamclu),main=NULL)
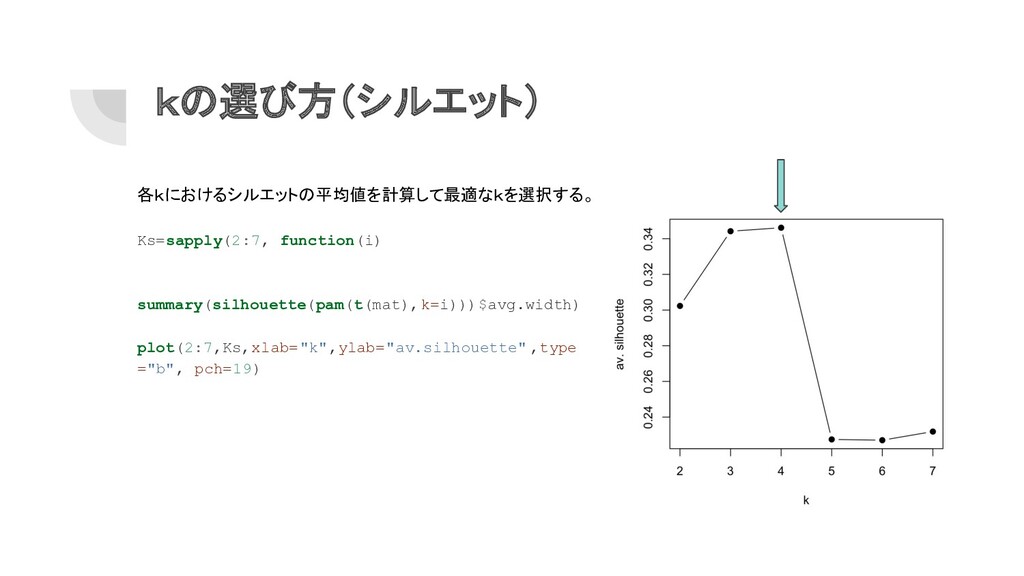
kの選び方(シルエット) 各kにおけるシルエットの平均値を計算して最適なkを選択する。 Ks=sapply(2:7, function(i) summary(silhouette(pam(t(mat),k=i)))$avg.width) plot(2:7,Ks,xlab="k",ylab="av.silhouette" ,type ="b", pch=19)
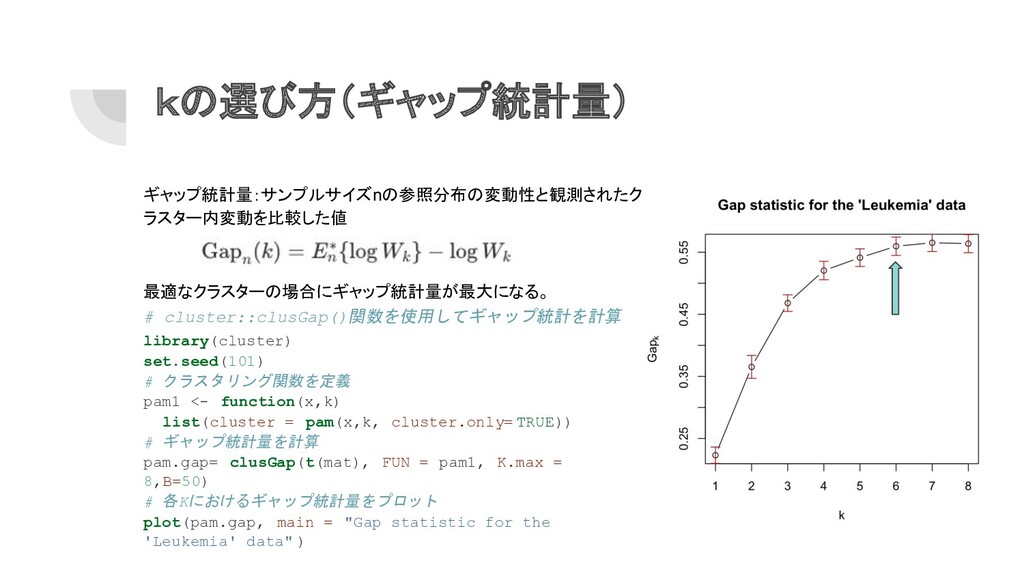
kの選び方(ギャップ統計量) ギャップ統計量:サンプルサイズnの参照分布の変動性と観測されたク ラスター内変動を比較した値 最適なクラスターの場合にギャップ統計量が最大になる。 # cluster::clusGap()関数を使用してギャップ統計を計算 library(cluster) set.seed(101) # クラスタリング関数を定義
pam1 <- function(x,k) list(cluster = pam(x,k, cluster.only= TRUE)) # ギャップ統計量を計算 pam.gap= clusGap(t(mat), FUN = pam1, K.max = 8,B=50) # 各Kにおけるギャップ統計量をプロット plot(pam.gap, main = "Gap statistic for the 'Leukemia' data" )
次元削減
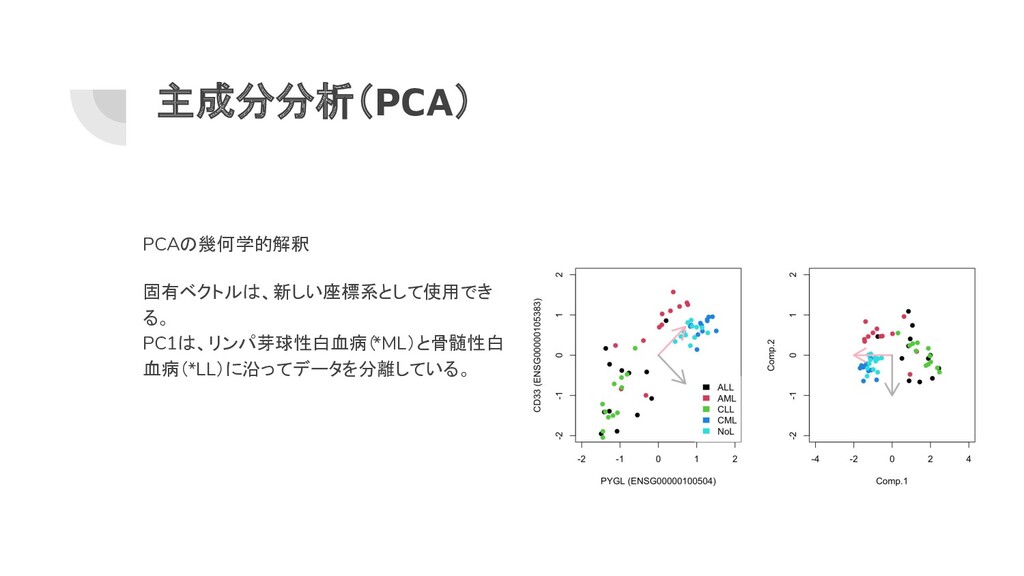
主成分分析(PCA) 高次元データを調べるための最も一般的な手法 新しい座標系の軸がデータの最大分散の方向 を指すように、元のデータ空間(座標)を回転さ せる。 最初のコンポーネントPC1 (Comp.1 ) は、デー タの分散が最も大きい方向を表す。
2番目のコンポーネントPC2 (Comp.2 ) は、最 初のコンポーネントに直交する残りの分散の最 大値を表す。 # 2つの遺伝子の発現量にPCAを適用 sub.mat=t(mat[rownames(mat) %in% c("ENSG00000100504","ENSG00000105383"),]) pr=princomp(scale(sub.mat)) pr ## Call: ## princomp(x = scale(sub.mat)) ## Standard deviations: ## Comp.1 Comp.2 ## 1.3378898 0.4203778 ## 2 variables and 60 observations.
主成分分析(PCA) PCAの幾何学的解釈 固有ベクトルは、新しい座標系として使用でき る。 PC1は、リンパ芽球性白血病( *ML)と骨髄性白 血病(*LL)に沿ってデータを分離している。
主成分分析(PCA) 固有値分解 PCAは、固有分解と呼ばれる操作を介して共分散行列の 固有ベクトルを計算することによって取得される。 # 共分散行列を計算 > cov.mat=cov(sub.mat) # 固有値分解の結果を取得(固有値、固有ベクトル)
> eigen(cov.mat) 固有ベクトルは方向を示し、固有値はその方向の変化を表 す。
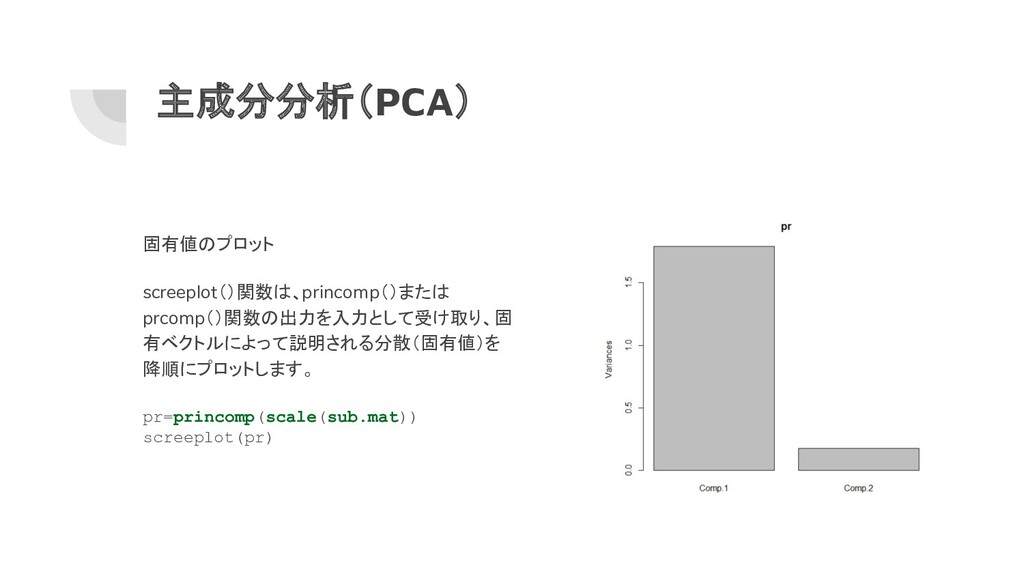
主成分分析(PCA) 固有値のプロット screeplot()関数は、princomp()または prcomp()関数の出力を入力として受け取り、固 有ベクトルによって説明される分散(固有値)を 降順にプロットします。 pr=princomp(scale(sub.mat)) screeplot(pr)
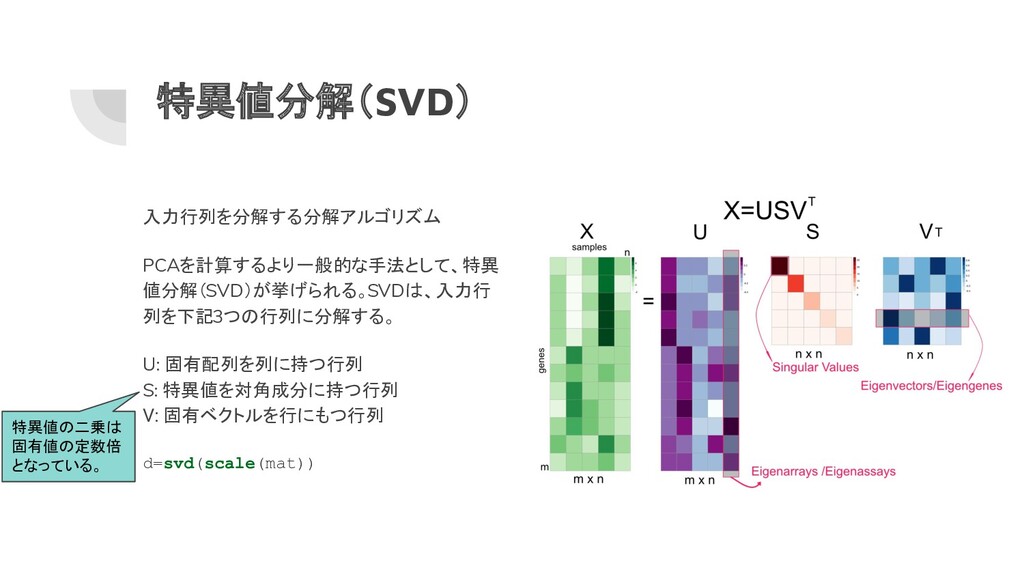
特異値分解(SVD) 入力行列を分解する分解アルゴリズム PCAを計算するより一般的な手法として、特異 値分解(SVD)が挙げられる。SVDは、入力行 列を下記3つの行列に分解する。 U: 固有配列を列に持つ行列 S: 特異値を対角成分に持つ行列 V:
固有ベクトルを行にもつ行列 d=svd(scale(mat)) 特異値の二乗は 固有値の定数倍 となっている。
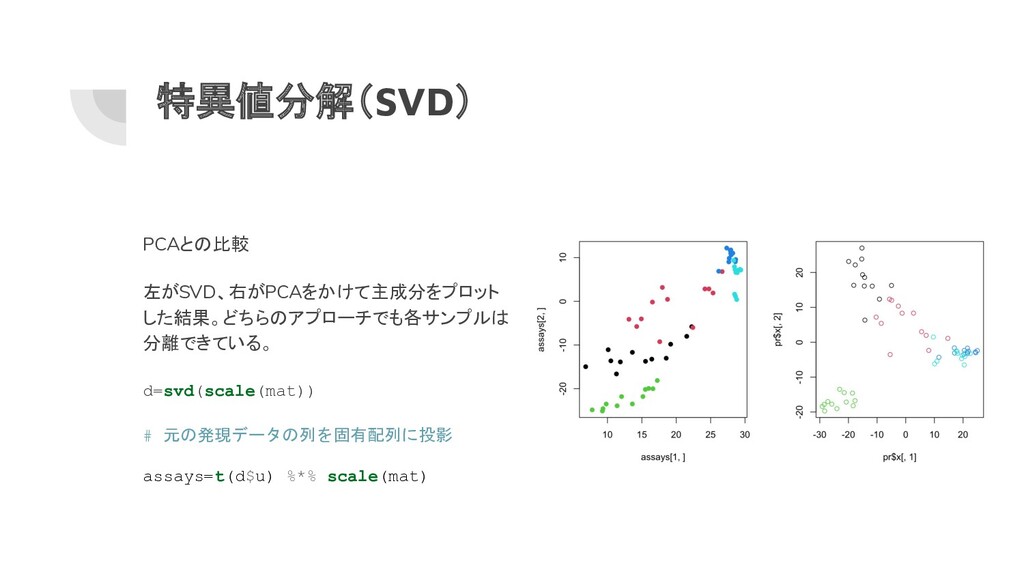
特異値分解(SVD) PCAとの比較 左がSVD、右がPCAをかけて主成分をプロット した結果。どちらのアプローチでも各サンプルは 分離できている。 d=svd(scale(mat)) # 元の発現データの列を固有配列に投影 assays=t(d$u) %*%
scale(mat)
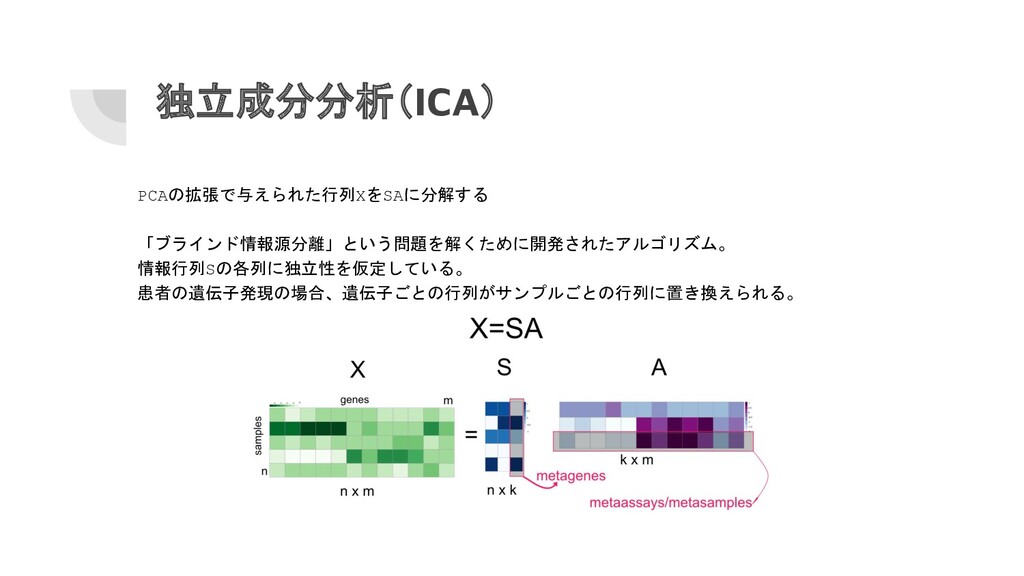
独立成分分析(ICA) PCAの拡張で与えられた行列XをSAに分解する 「ブラインド情報源分離」という問題を解くために開発されたアルゴリズム。 情報行列Sの各列に独立性を仮定している。 患者の遺伝子発現の場合、遺伝子ごとの行列がサンプルごとの行列に置き換えられる。
独立成分分析(ICA) fastICA()を使用して、2つのコンポーネントを抽出 して視覚化する。 library(fastICA) ica.res=fastICA(t(mat),n.comp=2) # ICA適用 # 遺伝子間の関係をプロット plot(ica.res$S[,1],ica.res$S[,2],
col=as.factor(annotation_col $LeukemiaType) )
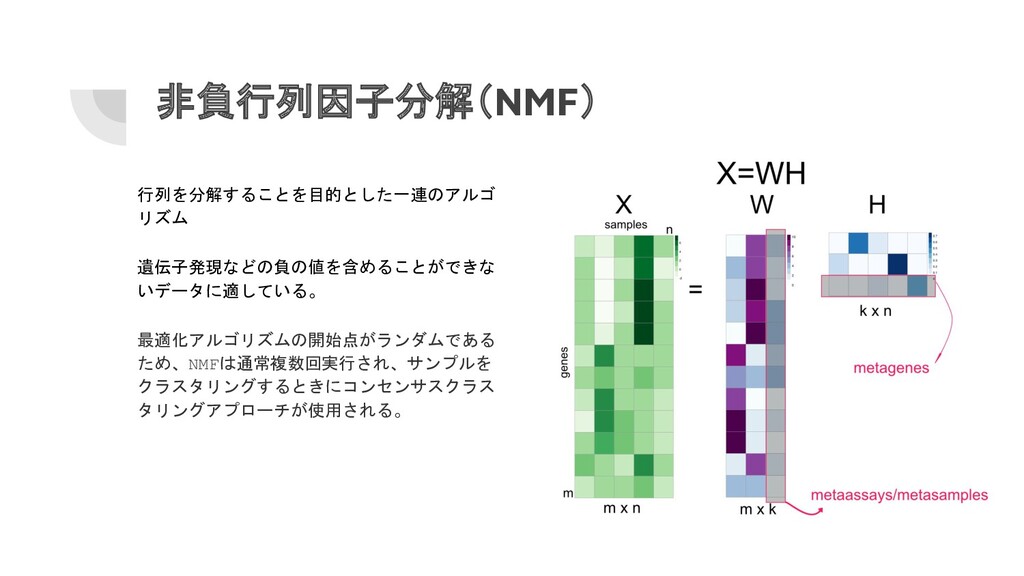
非負行列因子分解(NMF) 行列を分解することを目的とした一連のアルゴ リズム 遺伝子発現などの負の値を含めることができな いデータに適している。 最適化アルゴリズムの開始点がランダムである ため、NMFは通常複数回実行され、サンプルを クラスタリングするときにコンセンサスクラス タリングアプローチが使用される。
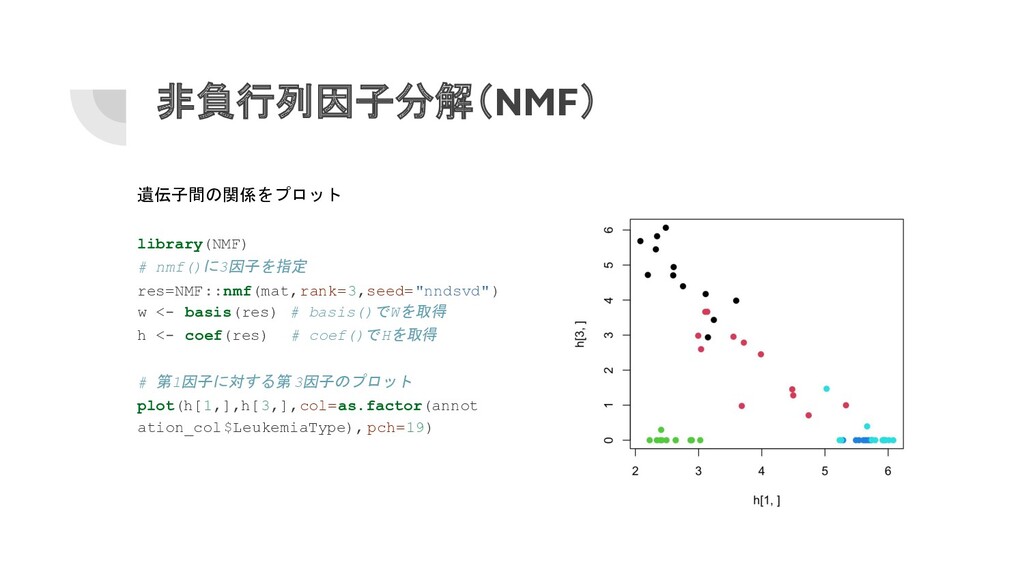
非負行列因子分解(NMF) 遺伝子間の関係をプロット library(NMF) # nmf()に3因子を指定 res=NMF::nmf(mat,rank=3,seed="nndsvd") w <- basis(res) #
basis()でWを取得 h <- coef(res) # coef()でHを取得 # 第1因子に対する第 3因子のプロット plot(h[1,],h[3,],col=as.factor(annot ation_col$LeukemiaType), pch=19)
多次元尺度構成法(MDS) 高次元空間の距離データの情報をあまり失うことなく 低次元空間に表示するデータ分析手法 非計量MDS:従来のMDSを改善し、低次元の距離が高次 元の距離にどの程度対応するかを再度測定する。 1. ランダムな低次元構成を見つけるか、従来の MDSに よって返される構成から始める。 2.
低次元のポイント間の距離を計算する。 3. 入力距離の最適な単調変換を見つける。 4. 低次元空間を再構成し、維持することにより、 Stress関 数を最小限に抑える。 5. 手順2から収束するまで繰り返す。
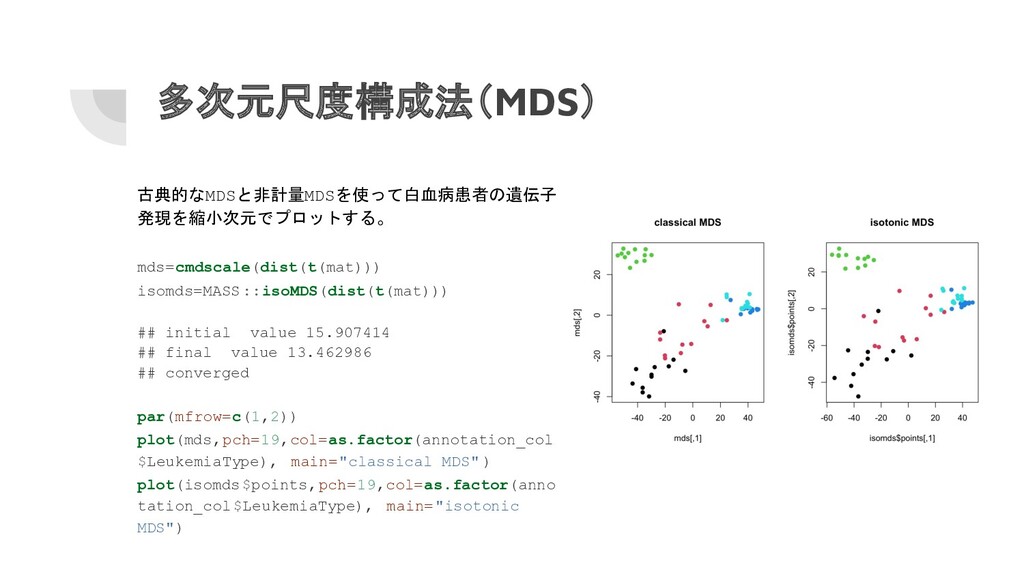
多次元尺度構成法(MDS) 古典的なMDSと非計量MDSを使って白血病患者の遺伝子 発現を縮小次元でプロットする。 mds=cmdscale(dist(t(mat))) isomds=MASS ::isoMDS(dist(t(mat))) ## initial value 15.907414
## final value 13.462986 ## converged par(mfrow=c(1,2)) plot(mds,pch=19,col=as.factor(annotation_col $LeukemiaType), main="classical MDS" ) plot(isomds$points,pch=19,col=as.factor(anno tation_col$LeukemiaType), main="isotonic MDS")
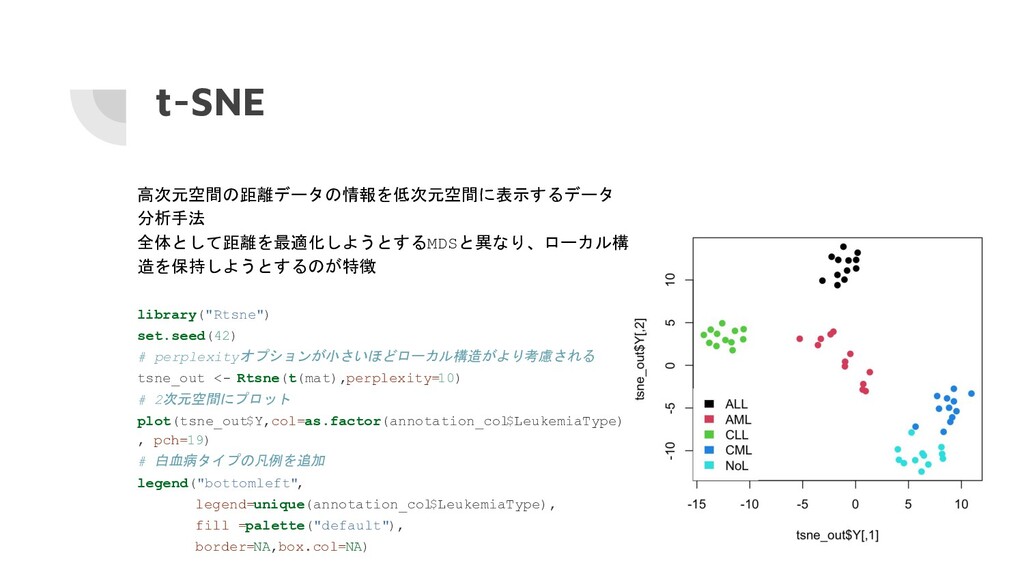
t-SNE 高次元空間の距離データの情報を低次元空間に表示するデータ 分析手法 全体として距離を最適化しようとするMDSと異なり、ローカル構 造を保持しようとするのが特徴 library("Rtsne") set.seed(42) # perplexityオプションが小さいほどローカル構造がより考慮される tsne_out
<- Rtsne(t(mat),perplexity=10) # 2次元空間にプロット plot(tsne_out$Y,col=as.factor(annotation_col $LeukemiaType) , pch=19) # 白血病タイプの凡例を追加 legend("bottomleft", legend=unique(annotation_col $LeukemiaType), fill =palette("default"), border=NA,box.col=NA)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![独立成分分析(ICA) fastICA()を使用して、2つのコンポーネントを抽出 して視覚化する。 library(fastICA) ica.res=fastICA(t(mat),n.comp=2) # ICA適用 # 遺伝子間の関係をプロット plot(ica.res$S[,1],ica.res$S[,2],](https://files.speakerdeck.com/presentations/e854b9a0a4f543c291906a72cdc4807c/slide_24.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}