Upgrade to Pro
— share decks privately, control downloads, hide ads and more …
Speaker Deck
Features
Speaker Deck
PRO
Sign in
Sign up for free
Search
Search
DNA methylation analysis using bisulfite sequen...
Search
onouyek
February 07, 2021
Science
920
0
Share
Embed
Copy iframe code
Copy JS code
Copy link
Start on current slide
DNA methylation analysis using bisulfite sequencing data
【第10回】ゼロから始めるゲノム解析(R編)
onouyek
February 07, 2021
More Decks by onouyek
See All by onouyek
Finding Open Reading Frames
onouyek
0
400
Inferring mRNA from Protein: Products and Reductions of Lists
onouyek
0
380
Finding the Longest Shared Subsequence: Finding K-mers, Writing Functions, and Using Binary Search
onouyek
0
300
Find a Motif in DNA: Exploring Sequence Similarity
onouyek
0
350
Finding the Hamming Distance: Counting Point Mutations
onouyek
0
470
Creating the Fibonacci Sequence: Writing, Testing, and Benchmarking Algorithms
onouyek
1
500
Transcribing DNA into mRNA: Mutating Strings, Reading and Writing Files
onouyek
0
560
Operations on Genomic Intervals and Genome Arithmetic
onouyek
0
370
Exploratory Data Analysis with Unsupervised Machine Learning
onouyek
0
320
Other Decks in Science
See All in Science
俺たちは本当に分かり合えるのか? ~ PdMとスクラムチームの “ずれ” を科学する
bonotake
2
2.5k
力学系から見た現代的な機械学習
hanbao
4
4.3k
[NLP2026 参加報告会] AI for Science まとめ / NLP2026
lychee1223
0
1.9k
「念のためのログ保存」を組織全体でやめるためのポリシーと仕組み作り
i2tsuki
2
190
イロレーティングを活用した関東大学サッカーの定量的実力評価 / A quantitative performance evaluation of Kanto University Football Association using Elo rating
konakalab
0
290
白金鉱業Vol.21【初学者向け発表枠】身近な例から学ぶ数理最適化の基礎 / Learning the Basics of Mathematical Optimization Through Everyday Examples
brainpadpr
1
760
データベース06: SQL (3/3) 副問い合わせ
trycycle
PRO
1
1k
(CVPR2026) Back to Basics: Let Denoising Generative Models Denoise
shumpei777
0
210
機械学習 - ニューラルネットワーク入門
trycycle
PRO
0
1.1k
Inside the Mind of an LLM
baggiponte
0
200
Understanding CVP Waveforms: Interpretation and Clinical Implications in Anesthesiology
taka88
0
640
ダメな自分の育て方―性格タイプの「劣等機能」から理解するニガテ克服術
ppillc
0
200
Featured
See All Featured
Let's Do A Bunch of Simple Stuff to Make Websites Faster
chriscoyier
508
140k
Crafting Experiences
bethany
1
210
Why Our Code Smells
bkeepers
PRO
340
58k
Digital Projects Gone Horribly Wrong (And the UX Pros Who Still Save the Day) - Dean Schuster
uxyall
1
1.9k
[RailsConf 2023] Rails as a piece of cake
palkan
59
6.7k
Templates, Plugins, & Blocks: Oh My! Creating the theme that thinks of everything
marktimemedia
31
2.8k
KATA
mclloyd
PRO
35
15k
A better future with KSS
kneath
240
18k
Hiding What from Whom? A Critical Review of the History of Programming languages for Music
tomoyanonymous
3
890
Discover your Explorer Soul
emna__ayadi
2
1.2k
Breaking role norms: Why Content Design is so much more than writing copy - Taylor Woolridge
uxyall
0
340
Put a Button on it: Removing Barriers to Going Fast.
kastner
60
4.3k
Transcript
DNAメチル化解析(BS-seq) DNA methylation analysis using bisulfite sequencing data 【第10回】ゼロから始めるゲノム解析( R編)
onouyek@2021年2月5日
DNAメチル化とは • シトシンメチル化(5mC)は真核生物のゲノムにおける主要な塩基修飾の1つ • 遺伝子発現のエピジェネティック制御メカニズムの一部を担う • 可逆的ではあるが、そのほとんどは細胞分裂を通して安定的に維持される • ヒトゲノムには約2800万のCpGが存在し、60−80%はメチル化されている •
CpGの10%未満はCpGアイランドと呼ばれる CGリッチな領域に存在する • DNAメチル化はゲノム上で一様には分布しておらず CpGの密度に関連する https://www.abcam.co.jp/epigenetics/dna-methylation-a-guide-1
バイサルファイトシーケンシング • DNAメチル化を測定するための最も信頼性が高く一般的な方 法の1つ • 一塩基単位の解像度でDNAメチル化を測定できる • バイサルファイト変換によって、メチル化されていない CはTに 変換される
• C -> T変異をカウントしてメチル化塩基の割合を計算する • DNAメチル化のゲノムワイドな定量的測定値を取得できる https://www.abcam.co.jp/epigenetics/dna-methylation-a-guide-1
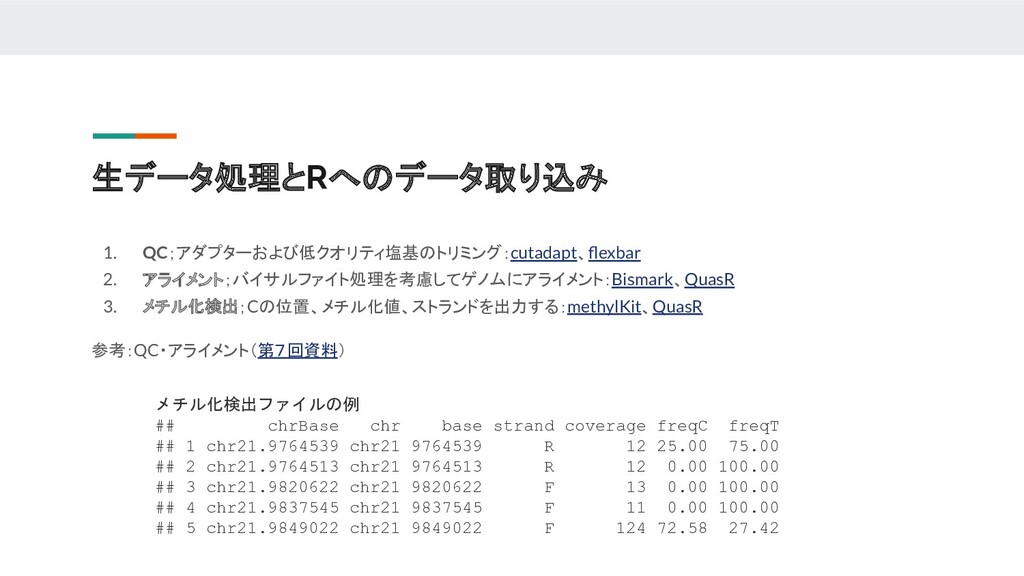
DNAメチル化解析フロー 1. 生データ処理 a. QC b. アライメント c. メチル化検出 d.
フィルタリング 2. 探索的解析 a. クラスタリング b. 主成分分析 3. メチル化変動解析 a. DMC/DMR b. メチル化セグメンテーション 4. アノテーション
生データ処理とRへのデータ取り込み 1. QC;アダプターおよび低クオリティ塩基のトリミング:cutadapt、flexbar 2. アライメント;バイサルファイト処理を考慮してゲノムにアライメント:Bismark、QuasR 3. メチル化検出;Cの位置、メチル化値、ストランドを出力する:methylKit、QuasR 参考:QC・アライメント(第7回資料) メチル化検出ファイルの例 ##
chrBase chr base strand coverage freqC freqT ## 1 chr21.9764539 chr21 9764539 R 12 25.00 75.00 ## 2 chr21.9764513 chr21 9764513 R 12 0.00 100.00 ## 3 chr21.9820622 chr21 9820622 F 13 0.00 100.00 ## 4 chr21.9837545 chr21 9837545 F 11 0.00 100.00 ## 5 chr21.9849022 chr21 9849022 F 124 72.58 27.42
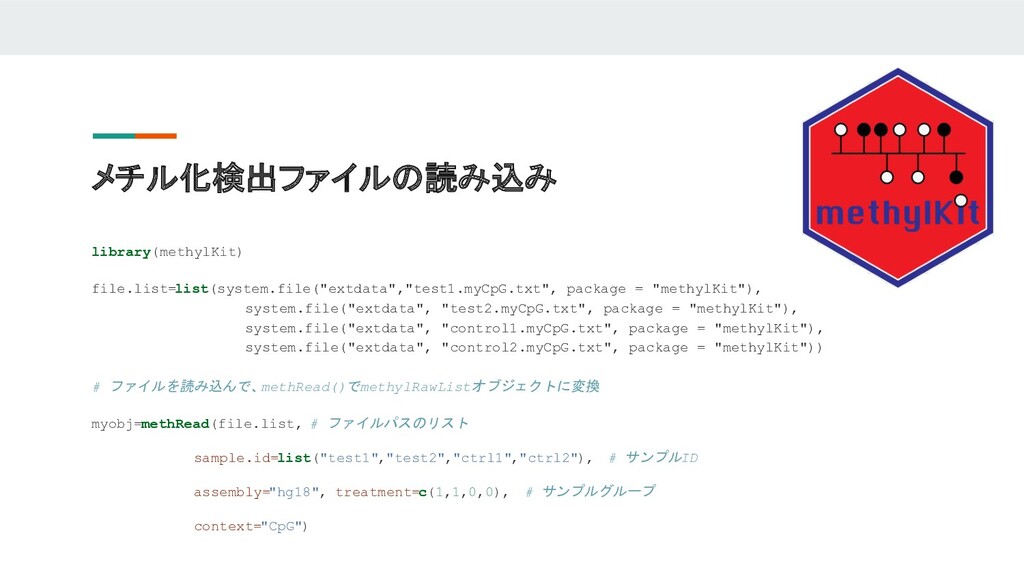
メチル化検出ファイルの読み込み library(methylKit) file.list=list(system.file("extdata","test1.myCpG.txt", package = "methylKit"), system.file("extdata", "test2.myCpG.txt", package =
"methylKit"), system.file("extdata", "control1.myCpG.txt", package = "methylKit"), system.file("extdata", "control2.myCpG.txt", package = "methylKit")) # ファイルを読み込んで、 methRead()でmethylRawListオブジェクトに変換 myobj=methRead(file.list, # ファイルパスのリスト sample.id=list("test1","test2","ctrl1","ctrl2"), # サンプルID assembly="hg18", treatment=c(1,1,0,0), # サンプルグループ context="CpG")
品質チェック(二峰性) CpGメチル化の二峰性の確認 getMethylationStats(myobj[[2]], plot=TRUE, both.strands= FALSE) ※二峰性からの大きな逸脱は実験の質が悪い可能性が疑われる
品質チェック(カバレッジ) CpGカバレッジの確認 getCoverageStats(myobj[[ 2]], plot=TRUE, both.strands= FALSE) ※カバレッジが異常に高い塩基は PCRバイアスの可能性が高い カバレッジがある一定の値より低い塩基と高い塩基を除外する
filtered.myobj= filterByCoverage(myobj, lo.count= 10, lo.perc= NULL, hi.count= NULL, hi.perc= 99.9)
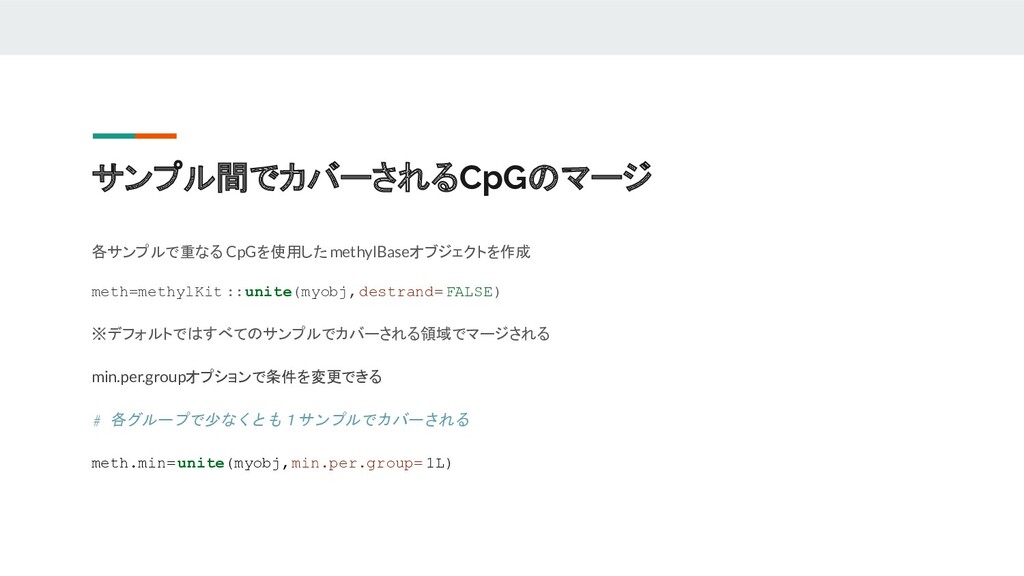
サンプル間でカバーされるCpGのマージ 各サンプルで重なる CpGを使用したmethylBaseオブジェクトを作成 meth=methylKit ::unite(myobj,destrand=FALSE) ※デフォルトではすべてのサンプルでカバーされる領域でマージされる min.per.groupオプションで条件を変更できる # 各グループで少なくとも1サンプルでカバーされる meth.min=unite(myobj,min.per.group=
1L)
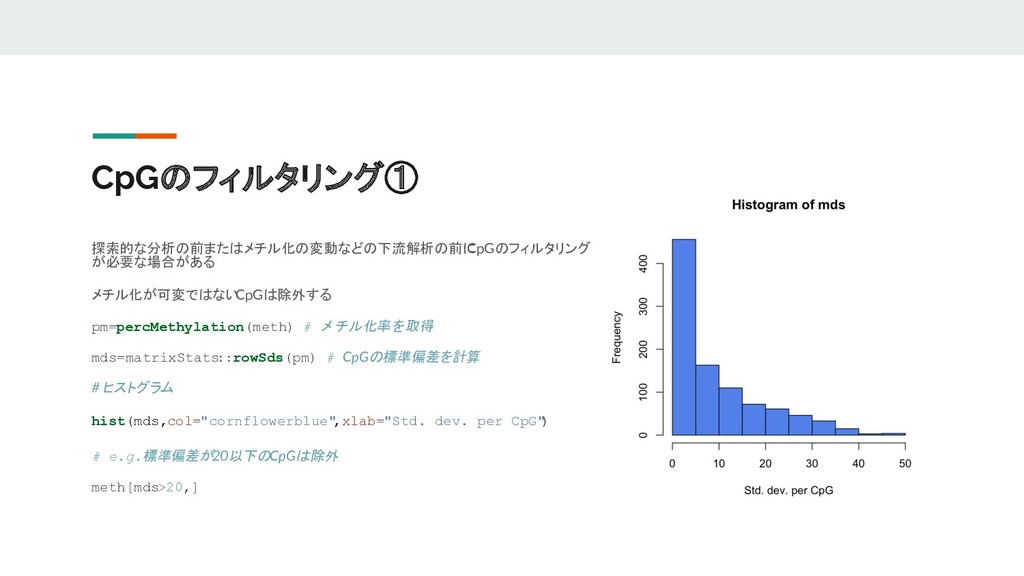
CpGのフィルタリング① 探索的な分析の前またはメチル化の変動などの下流解析の前に CpGのフィルタリング が必要な場合がある メチル化が可変ではない CpGは除外する pm=percMethylation(meth) # メチル化率を取得 mds=matrixStats
::rowSds(pm) # CpGの標準偏差を計算 # ヒストグラム hist(mds,col="cornflowerblue" ,xlab="Std. dev. per CpG" ) # e.g.標準偏差が20以下のCpGは除外 meth[mds>20,]
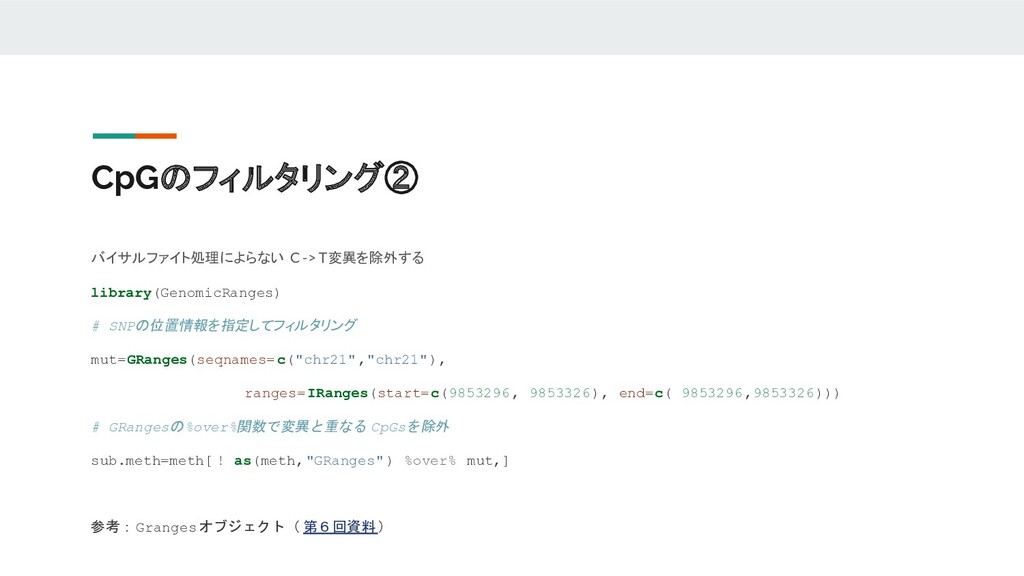
CpGのフィルタリング② バイサルファイト処理によらない C -> T変異を除外する library(GenomicRanges) # SNPの位置情報を指定してフィルタリング mut=GRanges(seqnames= c("chr21","chr21"),
ranges=IRanges(start=c(9853296, 9853326), end=c( 9853296,9853326))) # GRangesの%over%関数で変異と重なる CpGsを除外 sub.meth=meth[ ! as(meth,"GRanges" ) %over% mut,] 参考:Grangesオブジェクト( 第6回資料)
探索的解析
階層的クラスタリング 樹状図を描画して互いに類似したサンプルを見つける clusterSamples(meth, dist="correlation", # 相関距離 method="ward.D", # ウォード法 plot=TRUE)
参考:階層的クラスタリング( 第4回資料)
主成分分析(PCA) 散布図で互いに近いサンプルを確認する PCASamples(meth,adj.lim=c(1,1)) ※グループ内で離れている場合にはバッチ効果など実験 に問題がある可能性が疑われる 参考:PCA(第4回資料)
メチル化変動解析
メチル化変動解析(検定) グループ間でメチル化が変動した CpGサイト(DMC)および領域(DMR)を見つける フィッシャーの正確確率検定 :サンプルサイズが小さい場合に用いられる # グループ内のサンプルを 1つにまとめる pooled.meth= pool(meth,sample.ids=
c("test","control")) # 各グループに1サンプルずつの場合は自動的にフィッシャーの正確確率検定が適用される dm.pooledf= calculateDiffMeth(pooled.meth) ※グループ内のサンプル間変動は考慮されない
メチル化変動解析(抽出) 変動したメチル化 CpGを抽出する # 25%の差異と0.01のq値のcutoffを満たすすべてのメチル化変動塩基 /領域を抽出 all.diff=getMethylDiff(dm.pooledf, difference= 25,qvalue=0.01,type="all") #
25%の差異と0.01のq値のcutoffを満たすメチル化 増加塩基/領域を抽出 hyper=getMethylDiff(dm.pooledf, difference= 25,qvalue=0.01,type="hyper") # 25%の差異と0.01のq値のcutoffを満たすすべてのメチル化 減少塩基/領域を抽出 hypo=getMethylDiff(dm.pooledf, difference= 25,qvalue=0.01,type="hypo") # [ ]を使用してフィルタリング hyper2=dm.pooledf[dm.pooledf $qvalue < 0.01 & dm.pooledf$meth.diff > 25,]
メチル化変動解析(領域による要約) すべてのCpGがすべてのサンプルでカバーされているわけではない →領域を使用する tiles=tileMethylCounts(myobj,win.size=1000,step.size=1000) 特定の領域に関心がある場合(例:プロモーター) library(genomation) # BEDファイルから遺伝子の領域情報を読み込む gene.obj=readTranscriptFeatures(system.file("extdata","refseq.hg18.bed.txt" ,package="methylKit"
) ) # 関心領域をGRangesオブジェクトで指定する promoters=regionCounts(myobj,gene.obj $promoters)
メチル化変動解析(共変量の追加) 処置と共変量を含むモデルが共変量のみのモデルよりも優れているかをテストする covariates= data.frame(age=c(30,80,34,30,80,40)) # サンプルの年齢 # シュミレーションデータ sim.methylBase= dataSim(replicates= 6,sites=1000, treatment=c(rep(1,3),rep(0,3)),
covariates= covariates, sample.ids= c(paste0("test",1:3),paste0("ctrl",1:3))) # カイ二乗検定を使用したロジスティック回帰モデルを使用 my.diffMeth3= calculateDiffMeth(sim.methylBase, covariates= covariates, overdispersion= "MN", test="Chisq", mc.cores=1)
メチル化セグメンテーション サンプル内のメチル化プロファイリング methFile=system.file("extdata", "H1.chr21.chr22.rds" , package="compGenomRData" ) mbw=readRDS(methFile) res=methSeg(mbw, minSeg=10,
# セグメント内のCpG最小数 G=1:4, # 試行するセグメントクラスの数 join.neighbours = TRUE)
メチル化セグメンテーション 平均メチル化率とセグメントの長さの散布図をプロット plot(res$seg.mean, log10(width(res)),pch=20, col=scales::alpha(rainbow(4)[as.numeric(res$seg.group)], 0.2), ylab="log10(length)" , xlab="methylation proportion"
) メチル化率が高いセグメントは長くなる傾向がみられる
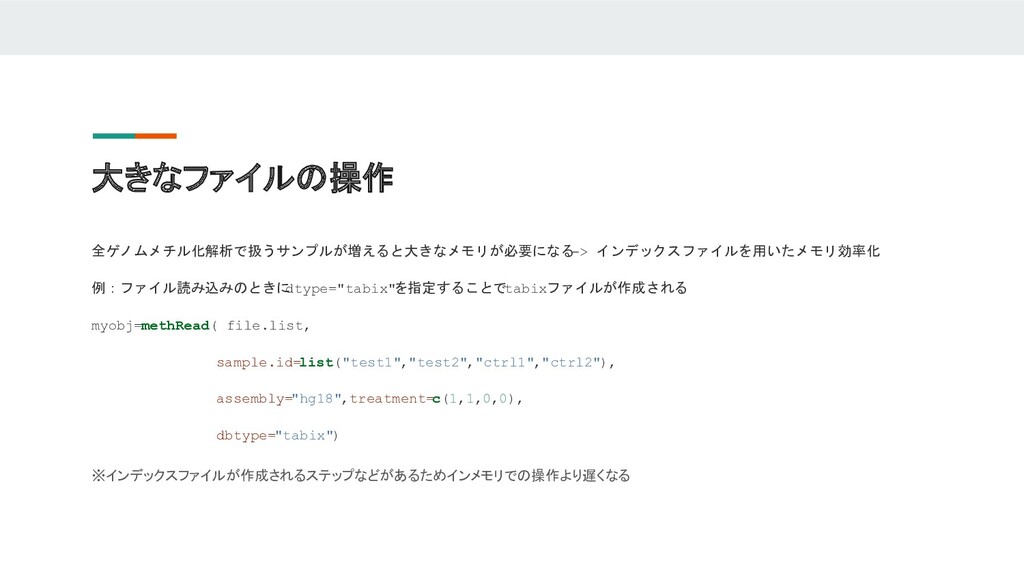
大きなファイルの操作 全ゲノムメチル化解析で扱うサンプルが増えると大きなメモリが必要になる -> インデックスファイルを用いたメモリ効率化 例:ファイル読み込みのときに dtype="tabix"を指定することでtabixファイルが作成される myobj=methRead( file.list, sample.id=list("test1","test2","ctrl1","ctrl2"), assembly="hg18",treatment=c(1,1,0,0),
dbtype="tabix") ※インデックスファイルが作成されるステップなどがあるためインメモリでの操作より遅くなる
アノテーション
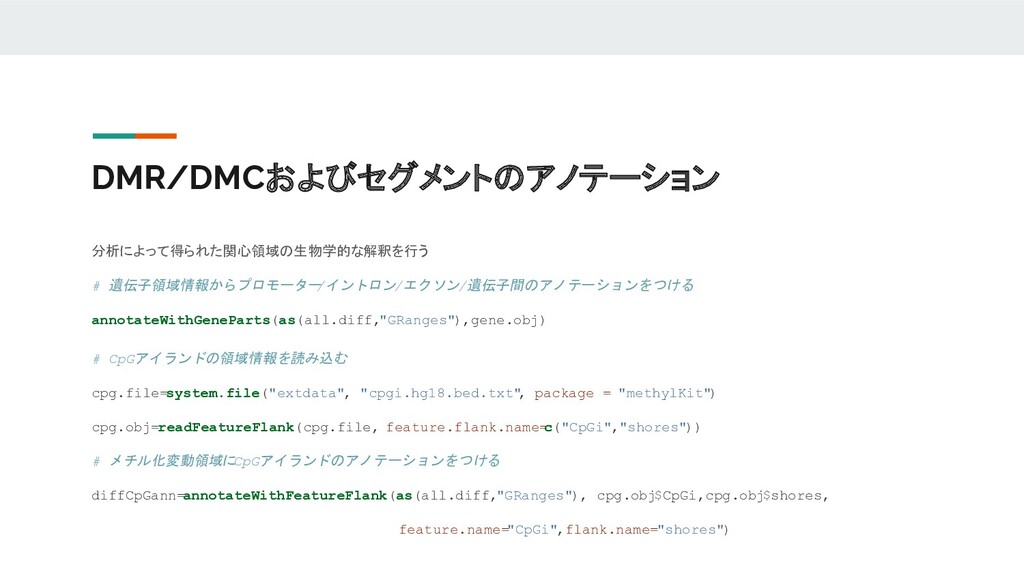
DMR/DMCおよびセグメントのアノテーション 分析によって得られた関心領域の生物学的な解釈を行う # 遺伝子領域情報からプロモーター /イントロン/エクソン/遺伝子間のアノテーションをつける annotateWithGeneParts(as(all.diff,"GRanges"),gene.obj) # CpGアイランドの領域情報を読み込む cpg.file=system.file("extdata", "cpgi.hg18.bed.txt"
, package = "methylKit") cpg.obj=readFeatureFlank(cpg.file, feature.flank.name= c("CpGi","shores")) # メチル化変動領域に CpGアイランドのアノテーションをつける diffCpGann=annotateWithFeatureFlank(as(all.diff,"GRanges"), cpg.obj$CpGi,cpg.obj$shores, feature.name="CpGi",flank.name="shores")
メチル化解析に使えるその他Rパッケージ DSS:経験ベイズを使用したベータ二項分布モデル BSseq:平滑化および線形回帰を使用したメチル化変動領域解析 BiSeq:ベータ二項分布モデルを使用したメチル化変動領域解析 MethylSeekR:HMMとcutoffを使用したメチロームセグメンテーション rGREAT:アノテーションおよびエンリッチメント解析 QuasR:BS-seqに対応したアライメント、メチル化検出および生データの QC
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![品質チェック(二峰性) CpGメチル化の二峰性の確認 getMethylationStats(myobj[[2]], plot=TRUE, both.strands= FALSE) ※二峰性からの大きな逸脱は実験の質が悪い可能性が疑われる](https://files.speakerdeck.com/presentations/522f79b96acf4306857f92b61e80b027/slide_6.jpg){kind=link}
![品質チェック(カバレッジ) CpGカバレッジの確認 getCoverageStats(myobj[[ 2]], plot=TRUE, both.strands= FALSE) ※カバレッジが異常に高い塩基は PCRバイアスの可能性が高い カバレッジがある一定の値より低い塩基と高い塩基を除外する](https://files.speakerdeck.com/presentations/522f79b96acf4306857f92b61e80b027/slide_7.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![メチル化セグメンテーション 平均メチル化率とセグメントの長さの散布図をプロット plot(res$seg.mean, log10(width(res)),pch=20, col=scales::alpha(rainbow(4)[as.numeric(res$seg.group)], 0.2), ylab="log10(length)" , xlab="methylation proportion"](https://files.speakerdeck.com/presentations/522f79b96acf4306857f92b61e80b027/slide_20.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}