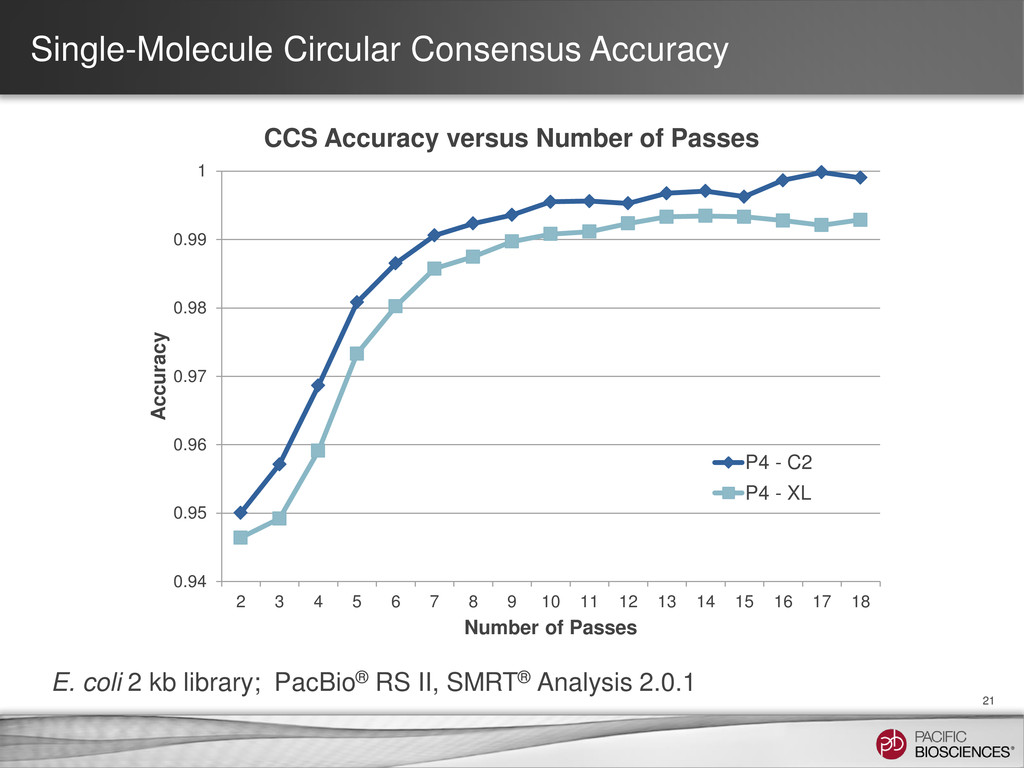
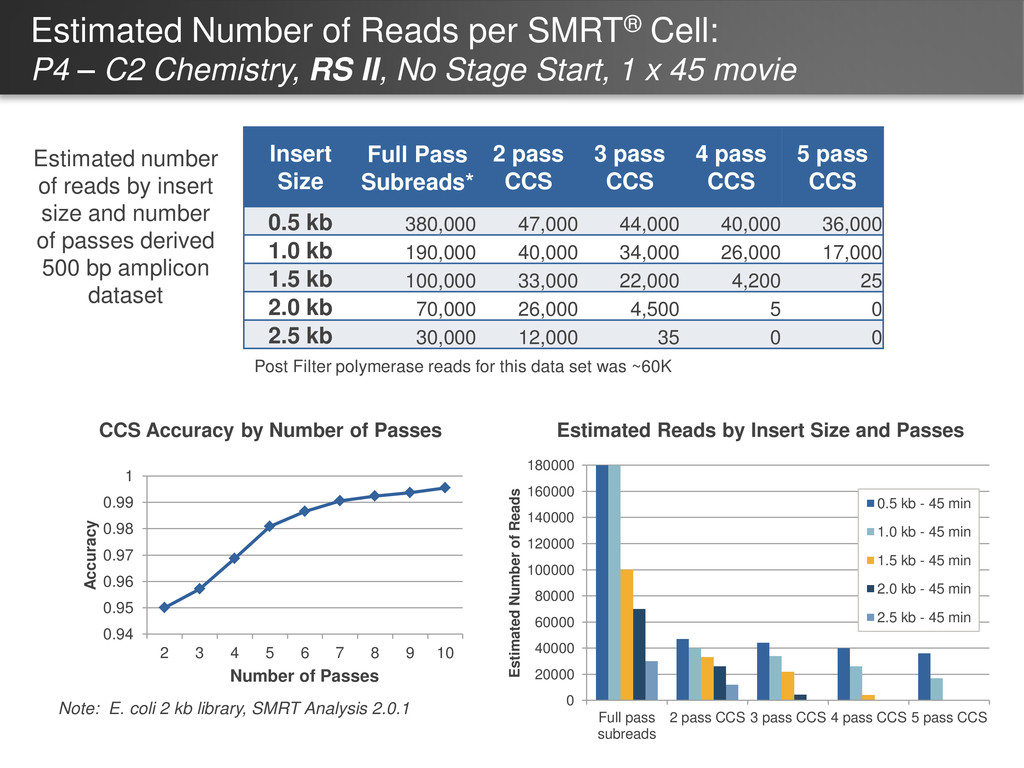
Chemistry, RS II, No Stage Start, 1 x 45 movie Insert Size Full Pass Subreads* 2 pass CCS 3 pass CCS 4 pass CCS 5 pass CCS 0.5 kb 380,000 47,000 44,000 40,000 36,000 1.0 kb 190,000 40,000 34,000 26,000 17,000 1.5 kb 100,000 33,000 22,000 4,200 25 2.0 kb 70,000 26,000 4,500 5 0 2.5 kb 30,000 12,000 35 0 0 Estimated number of reads by insert size and number of passes derived 500 bp amplicon dataset Note: E. coli 2 kb library, SMRT Analysis 2.0.1 Post Filter polymerase reads for this data set was ~60K 0.94 0.95 0.96 0.97 0.98 0.99 1 2 3 4 5 6 7 8 9 10 Accuracy Number of Passes CCS Accuracy by Number of Passes 0 20000 40000 60000 80000 100000 120000 140000 160000 180000 Full pass subreads 2 pass CCS 3 pass CCS 4 pass CCS 5 pass CCS Estimated Number of Reads 0.5 kb - 45 min 1.0 kb - 45 min 1.5 kb - 45 min 2.0 kb - 45 min 2.5 kb - 45 min Estimated Reads by Insert Size and Passes
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
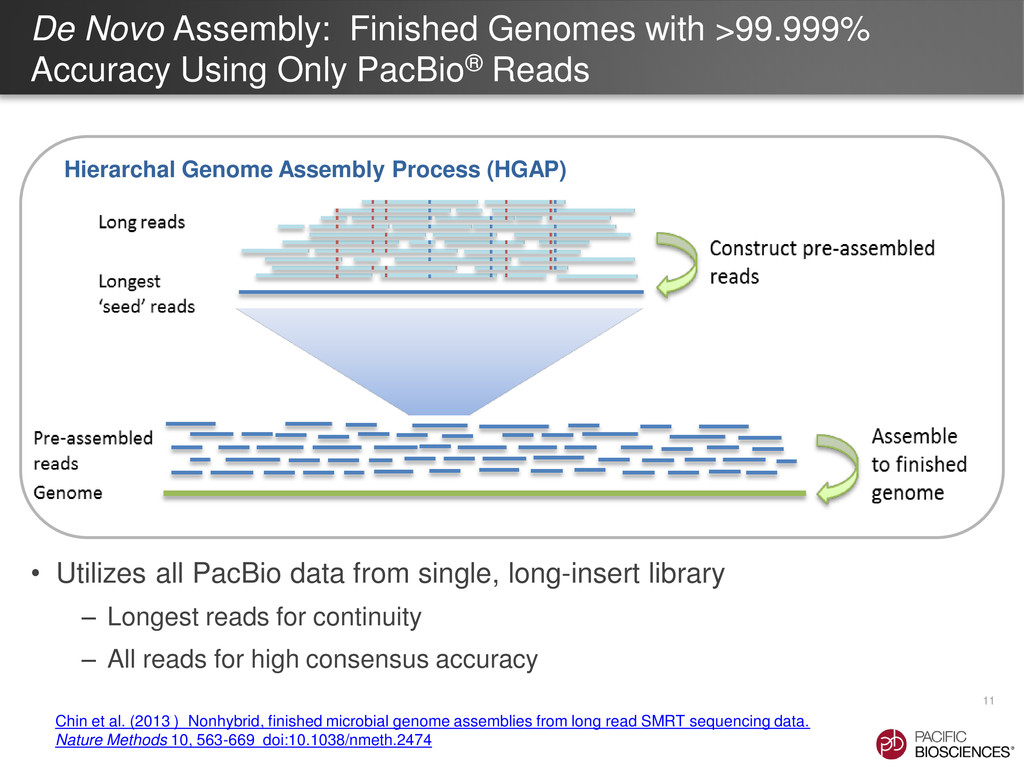{kind=link}
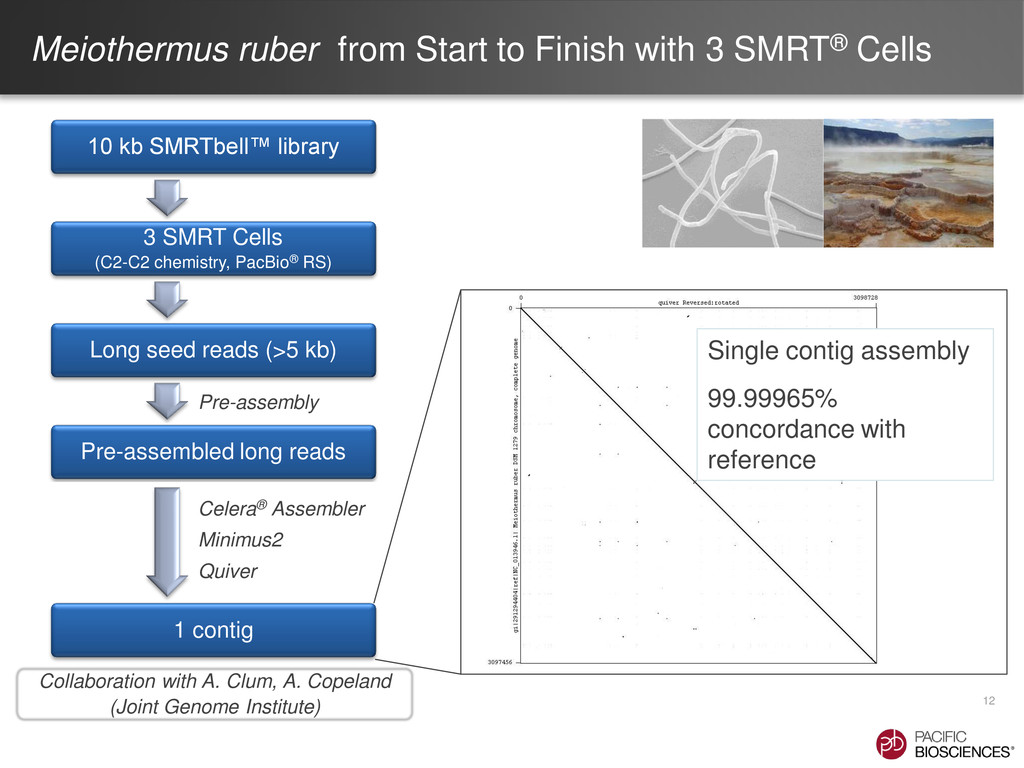{kind=link}
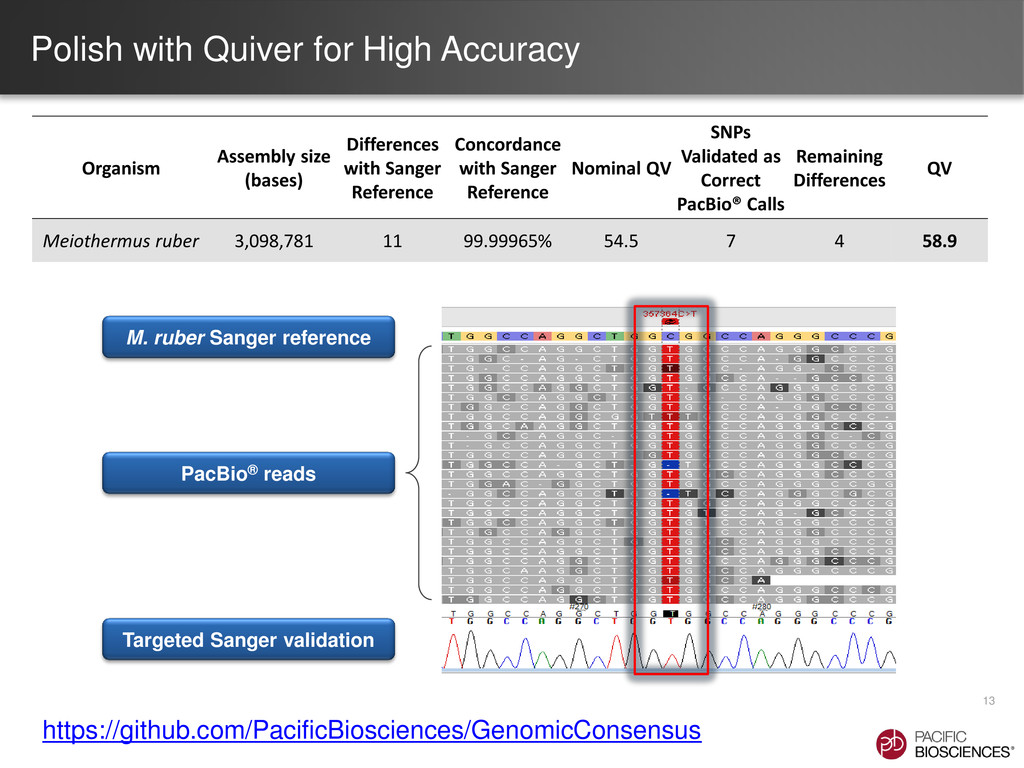{kind=link}
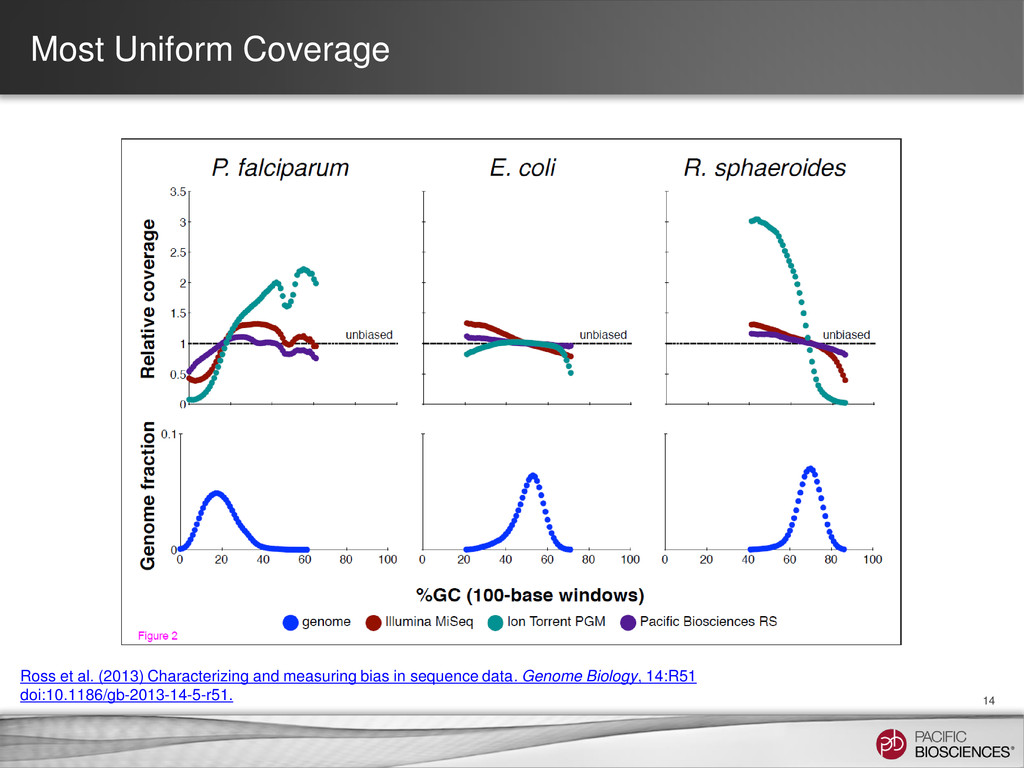{kind=link}
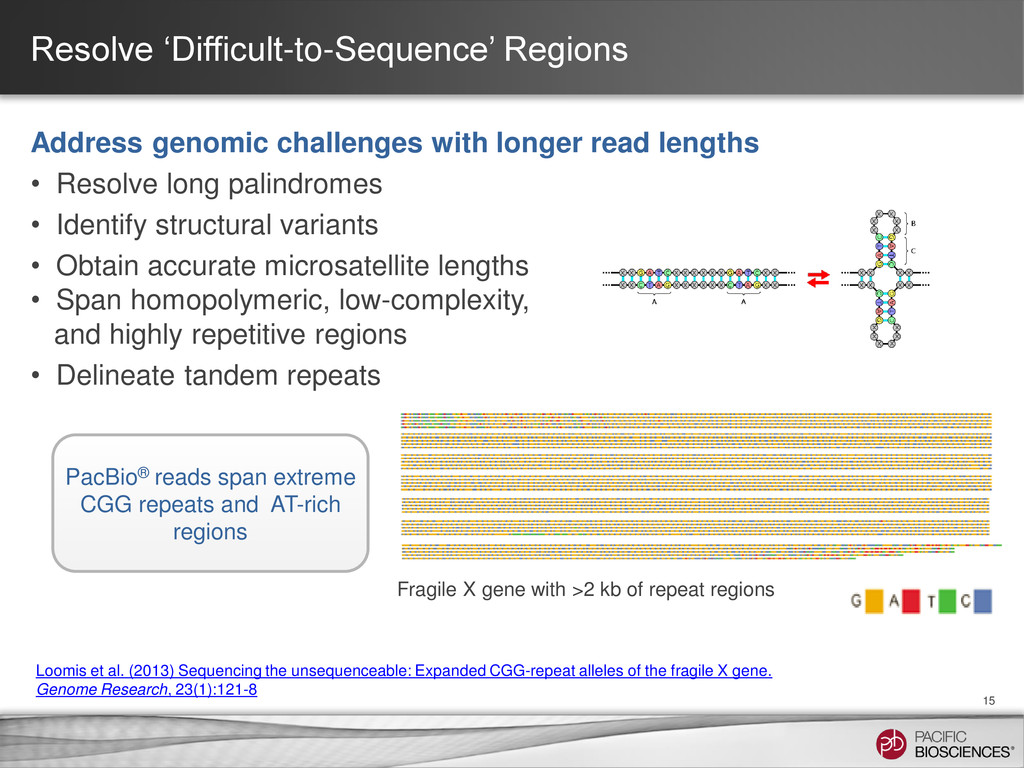{kind=link}
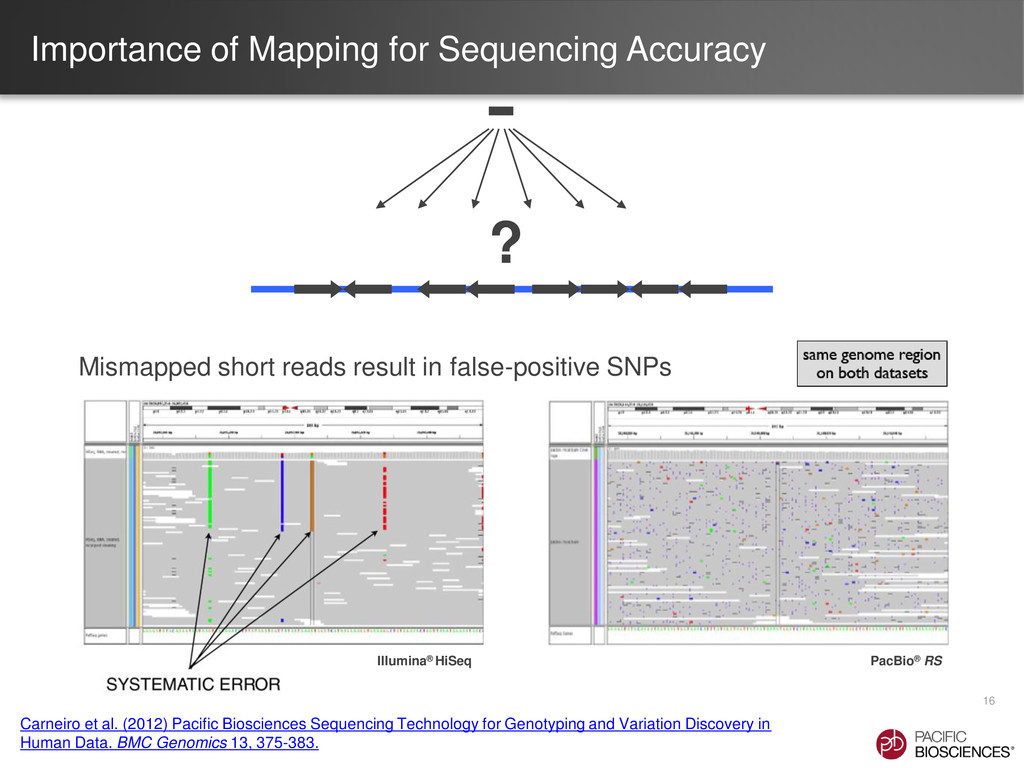{kind=link}
{kind=link}
{kind=link}
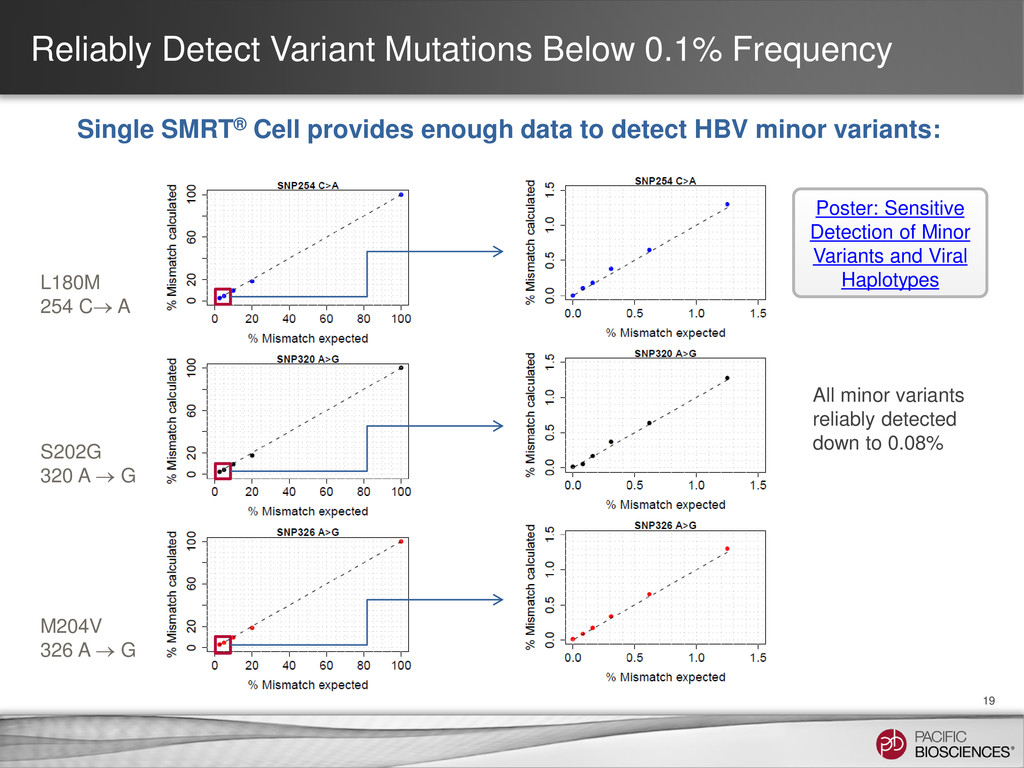{kind=link}
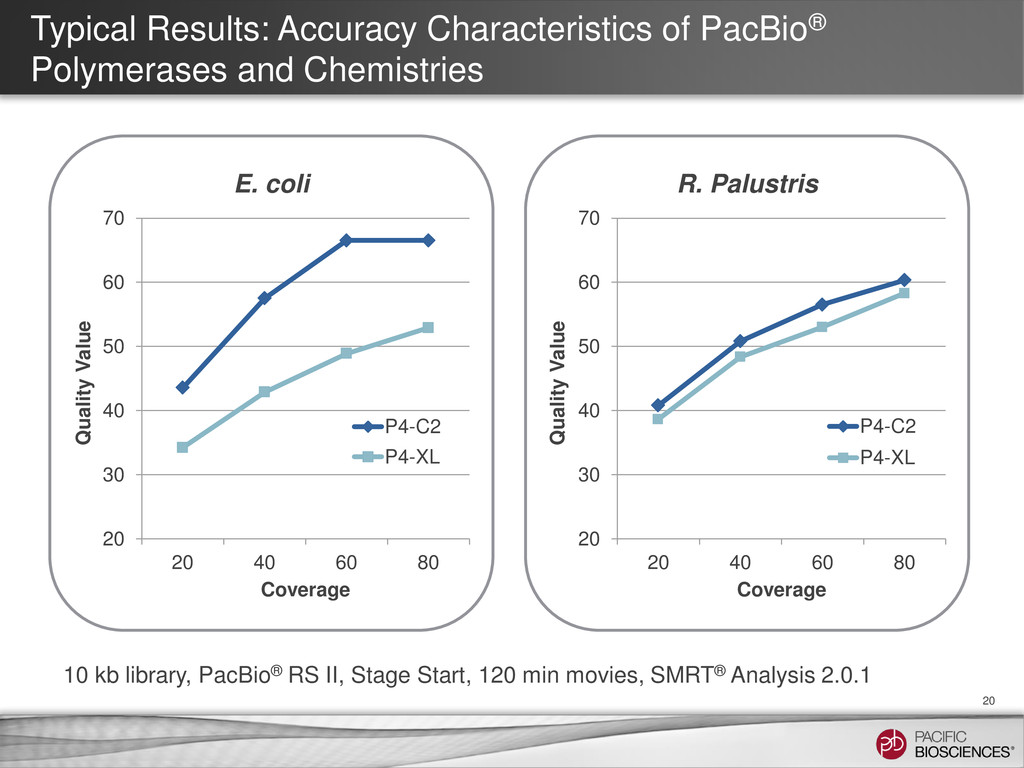{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
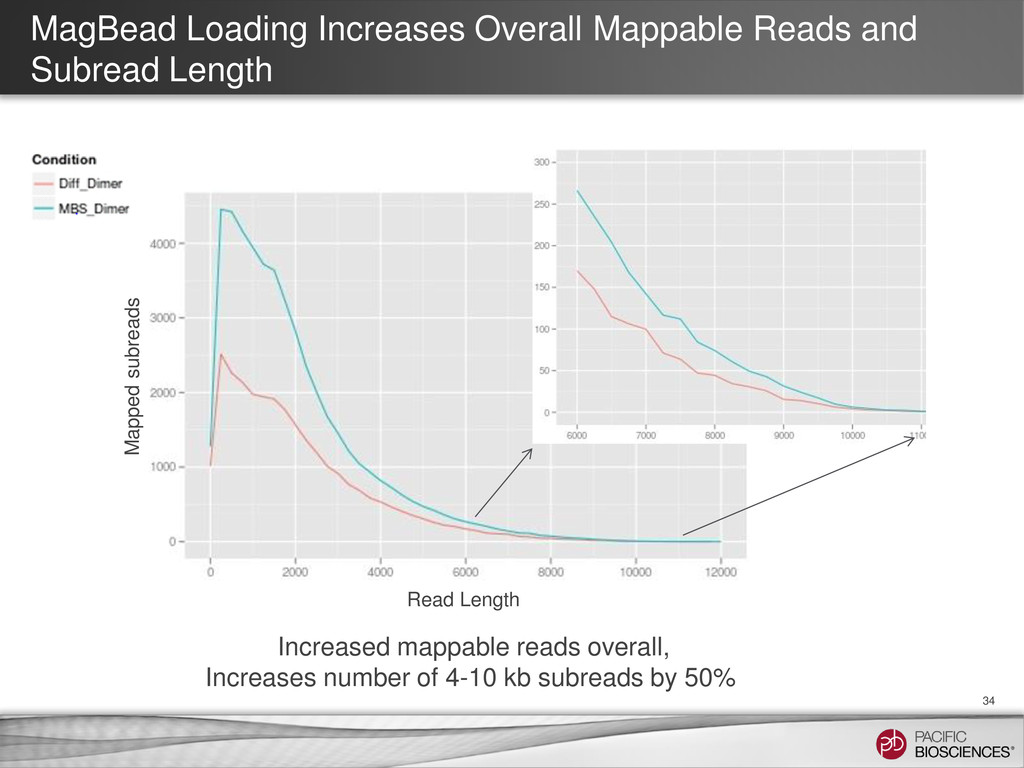{kind=link}
{kind=link}
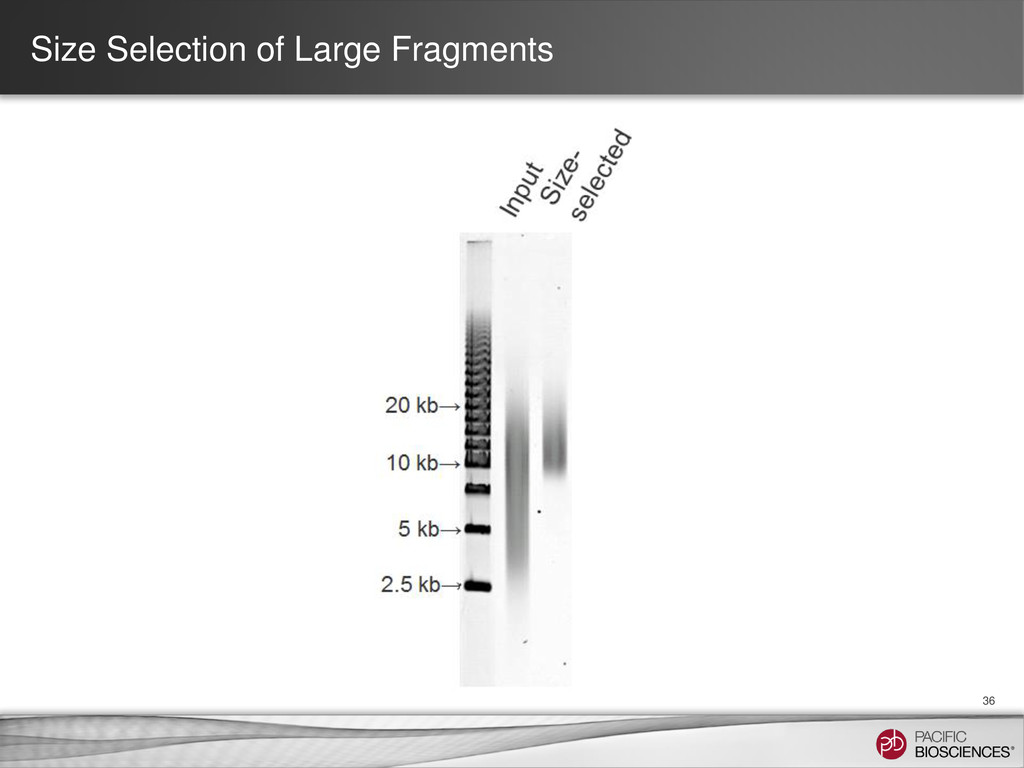{kind=link}
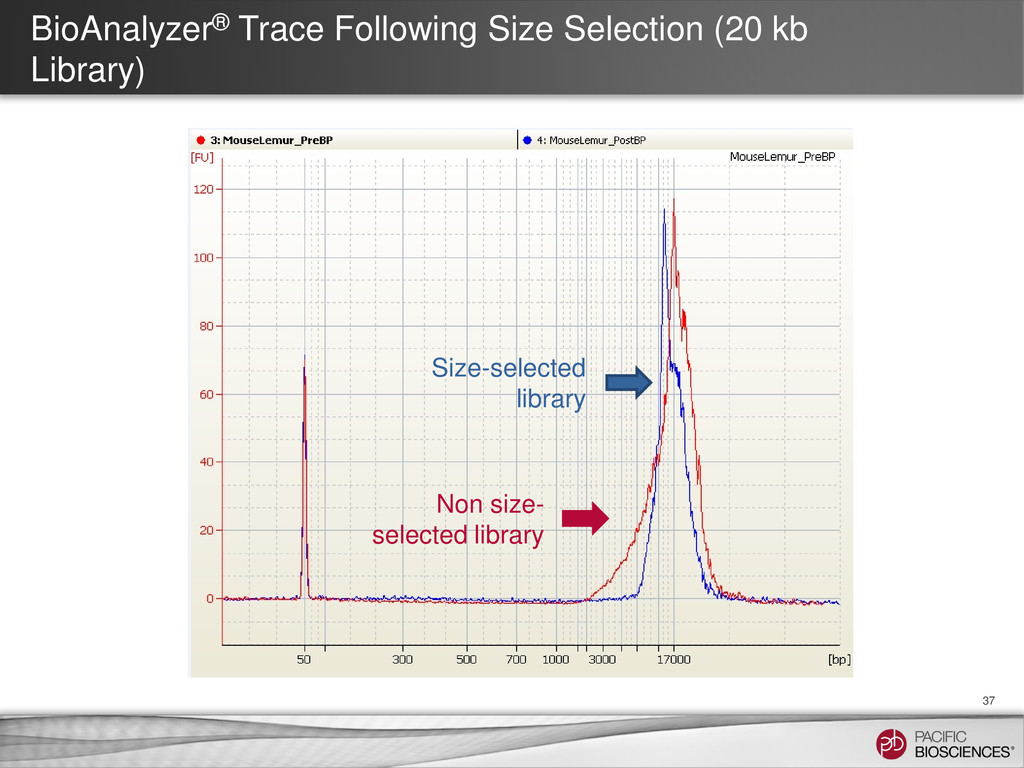{kind=link}
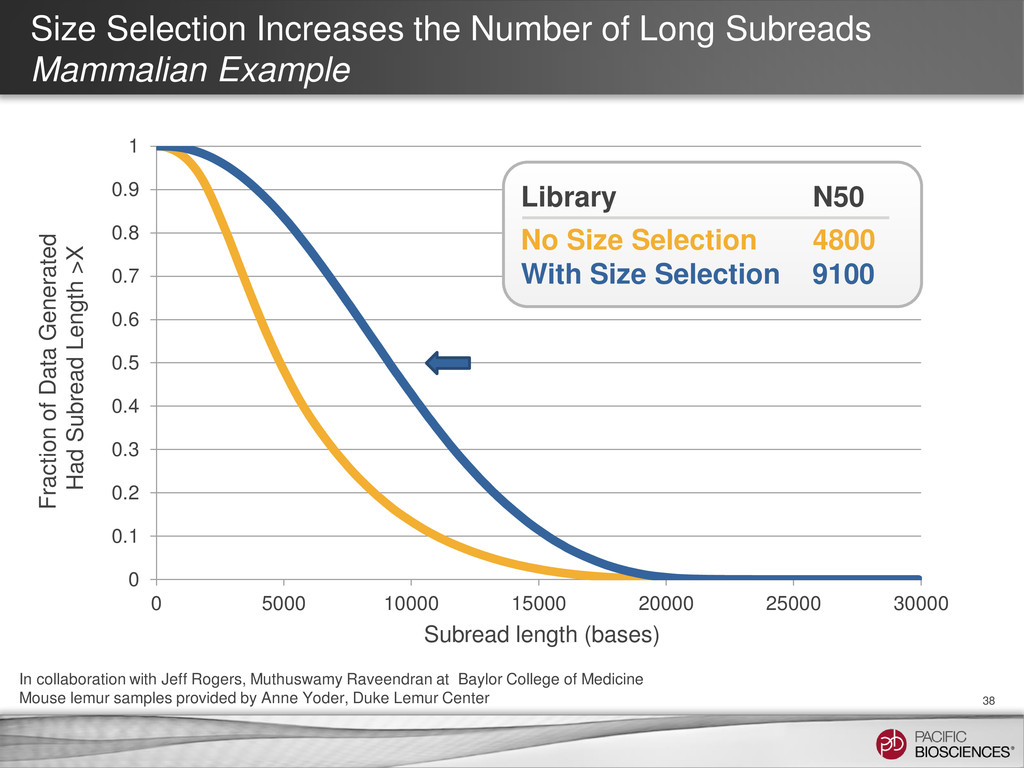{kind=link}
{kind=link}
{kind=link}
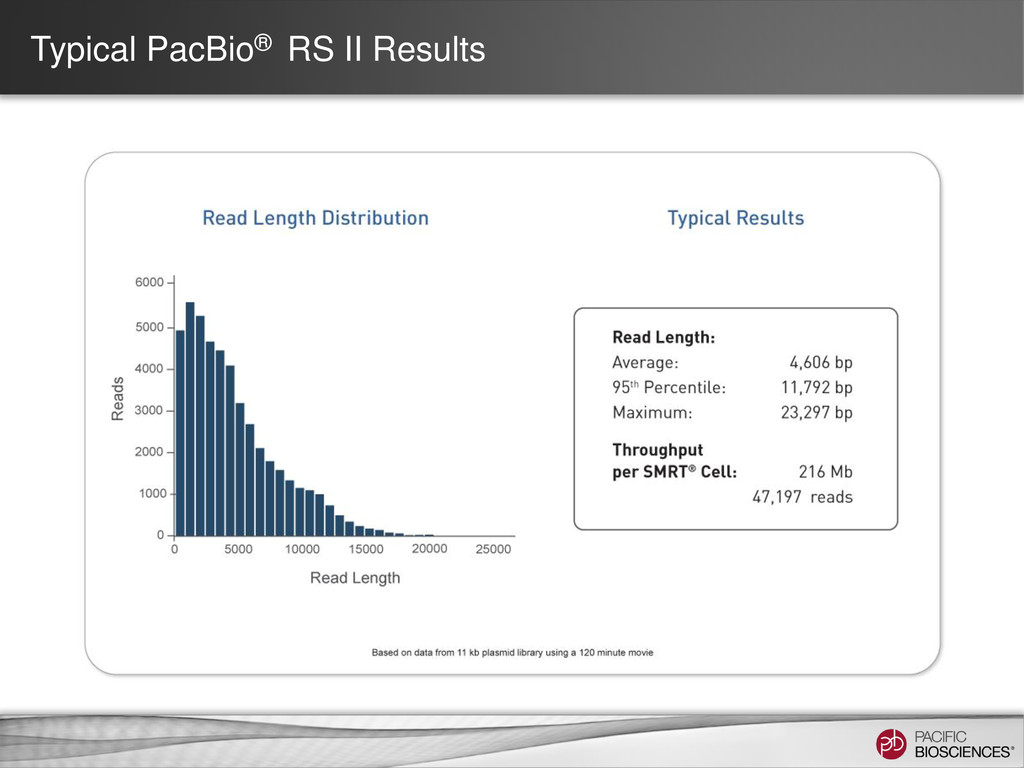{kind=link}
{kind=link}
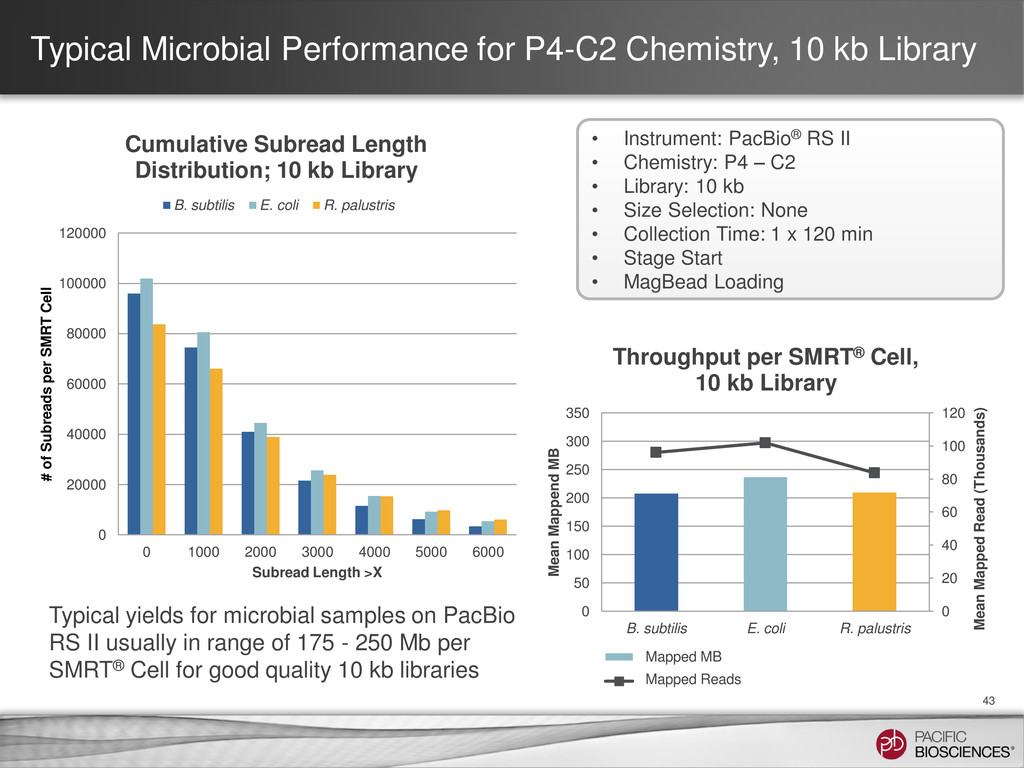{kind=link}
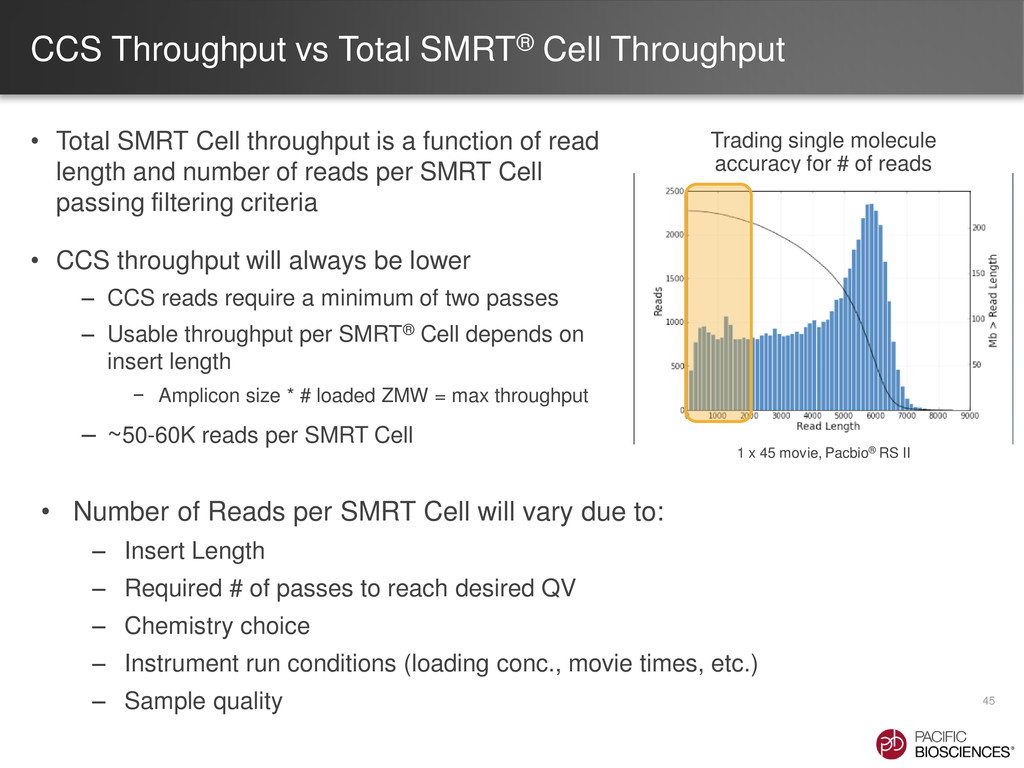{kind=link}
{kind=link}
{kind=link}
{kind=link}
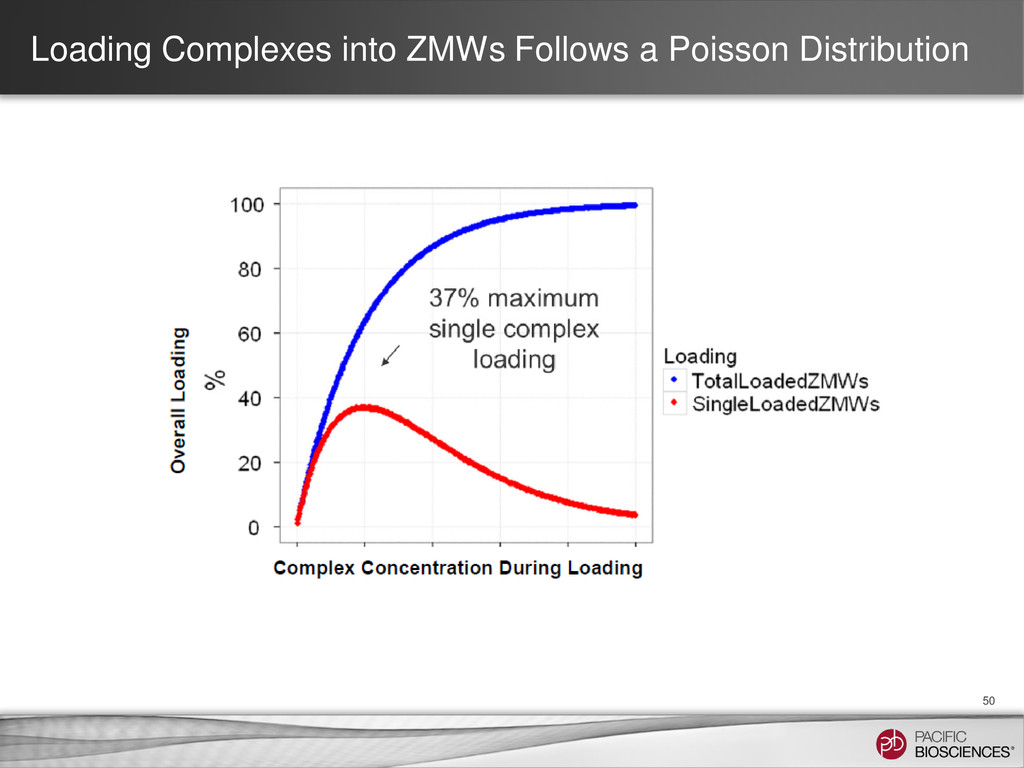{kind=link}
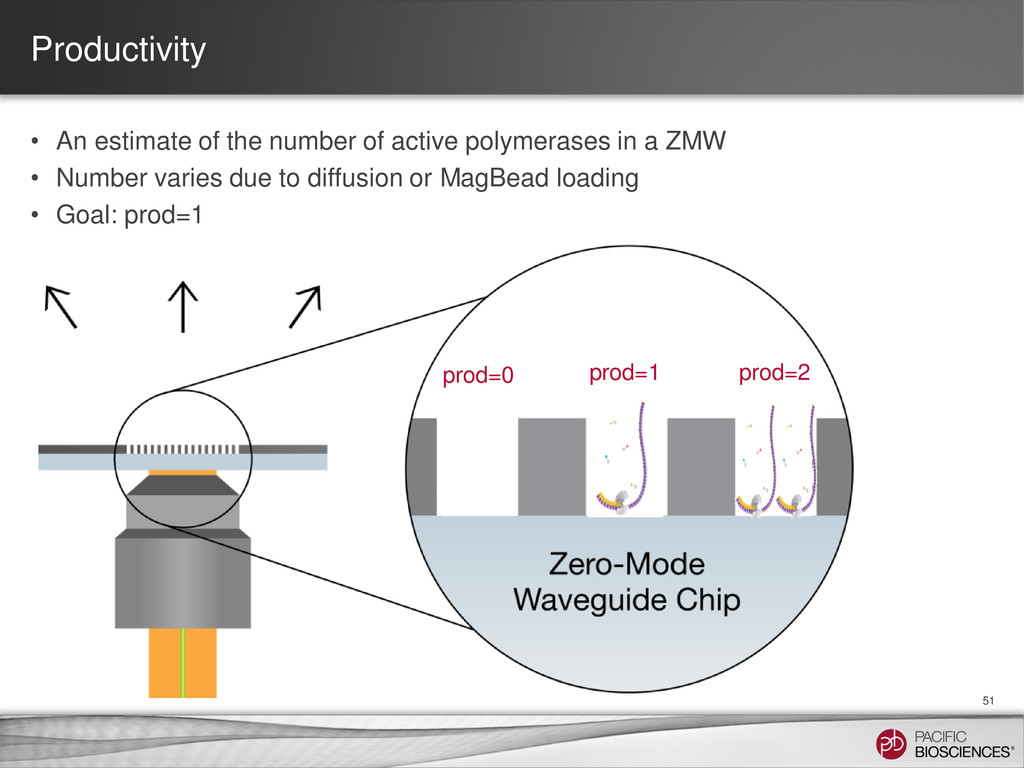{kind=link}
{kind=link}
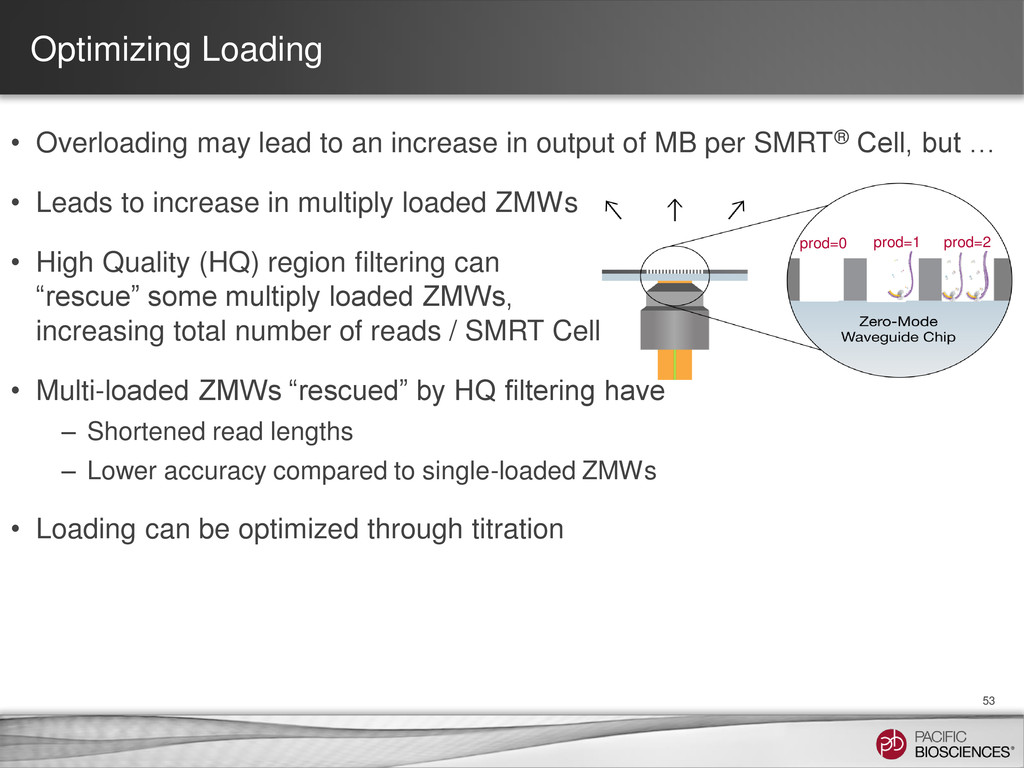{kind=link}
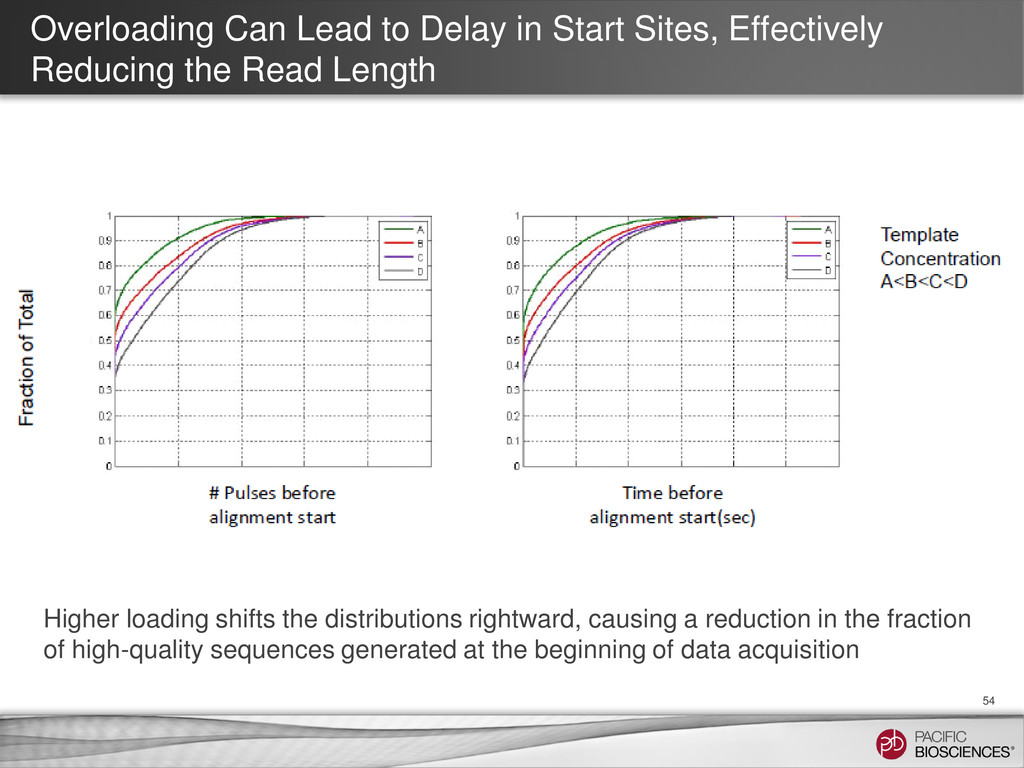{kind=link}
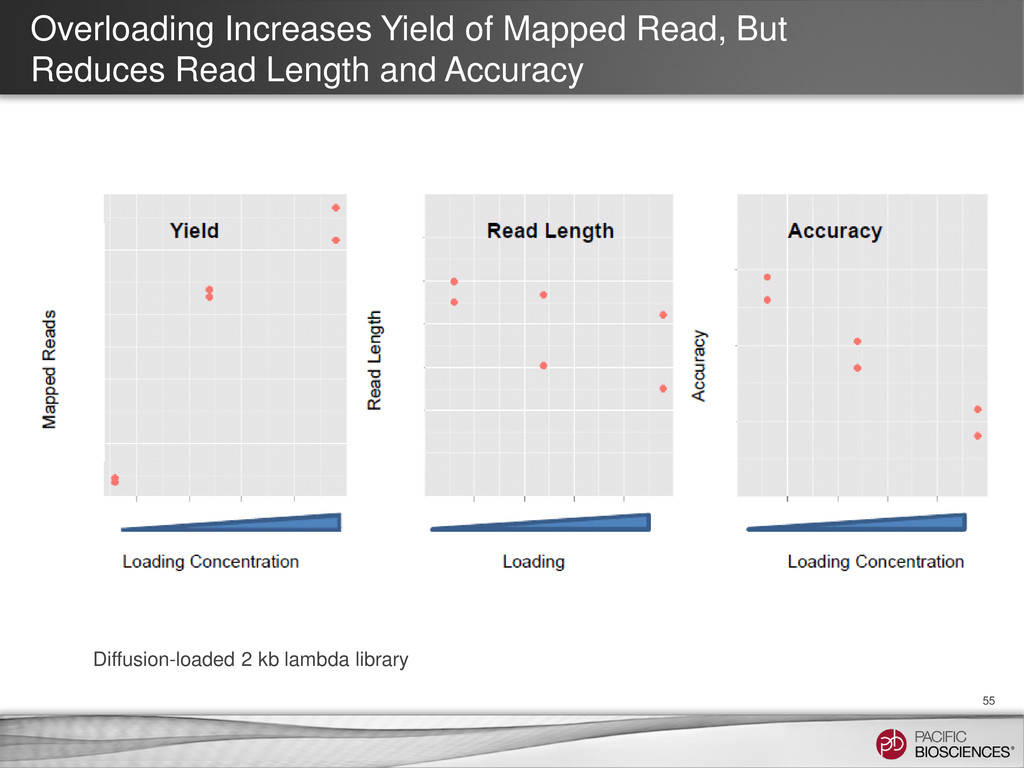{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}