now is… you take the transcriptome, you blow it up into pieces and then you try to figure out how they all go back together again… If you think about it, it’s kind of a crazy way to do things” Michael Synder Professor and Chair of Genetics Stanford University Tal Nawy, End to end RNA Sequencing, Nature Methods, v10, n10, Dec . 2013, p1144–1145 Ian Korf (2013) Genomics: the state of the art in RNA-seq analysis, Nature Methods, Nov 26;10(12):1165-6. doi: 10.1038/nmeth.2735.
(2013) Assessment of transcript reconstruction methods for RNA-Seq. Nature Methods doi:10.1038/nmeth.2714. …the complexity of higher eukaryotic genomes imposes severe limitations on transcript recall and splice product discrimination… …assembly of complete isoform structures poses a major challenge even when all constituent elements are identified… …Ultimately, the evolution of RNA-seq will move toward single- pass determination of intact transcripts….
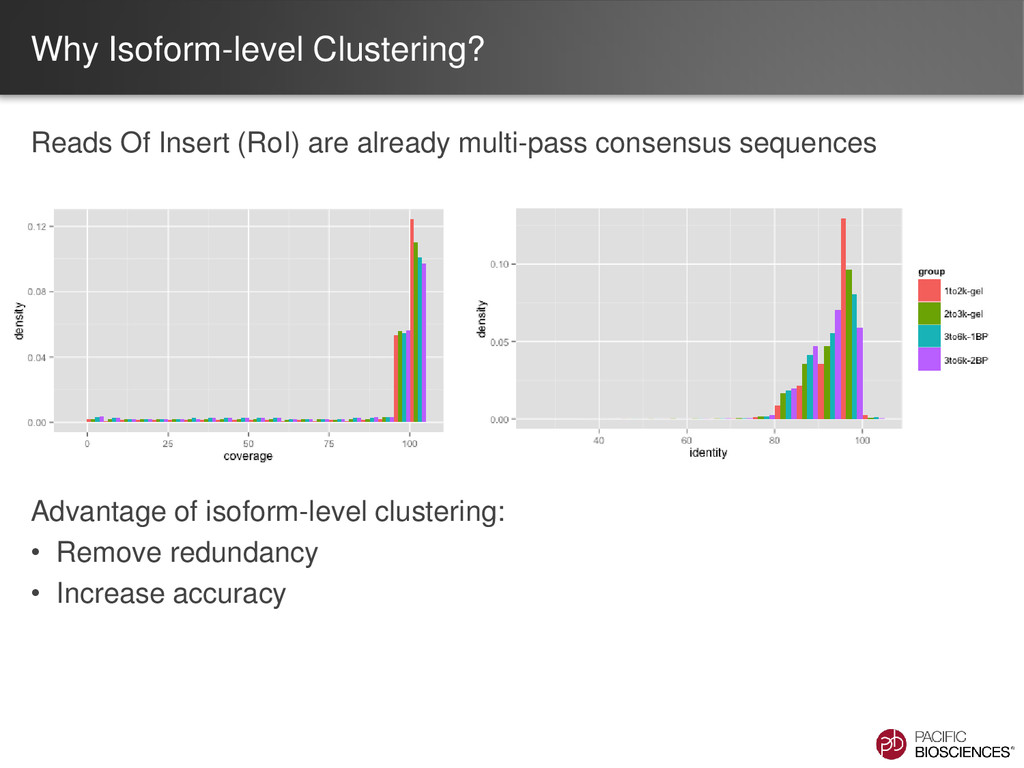
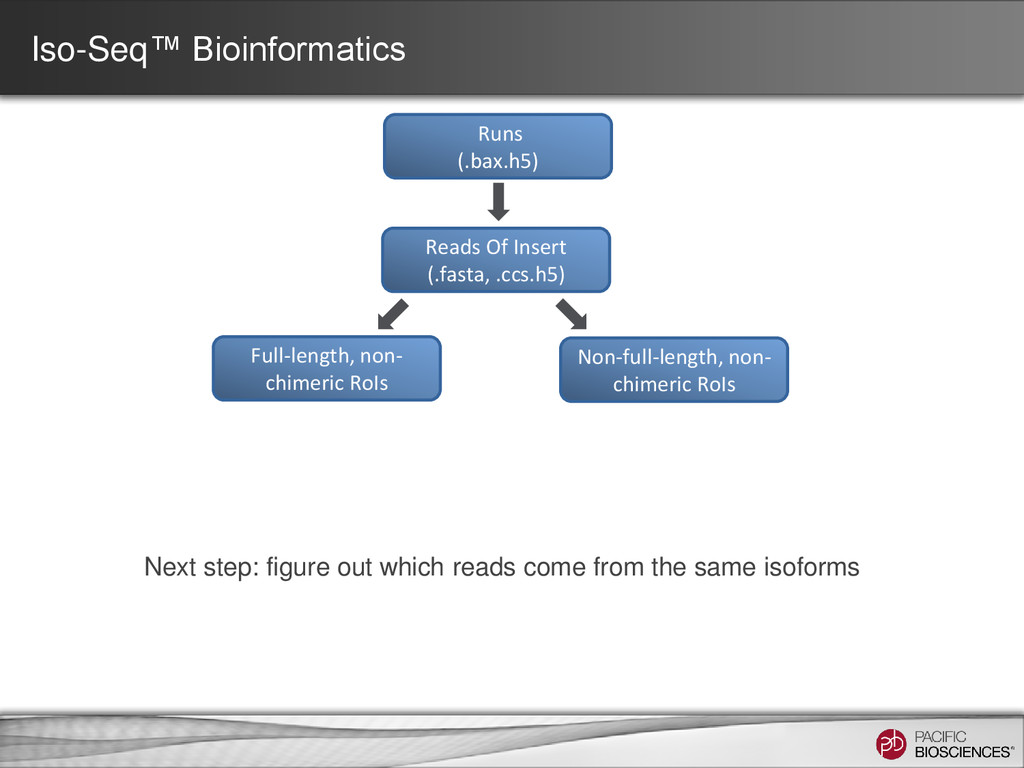
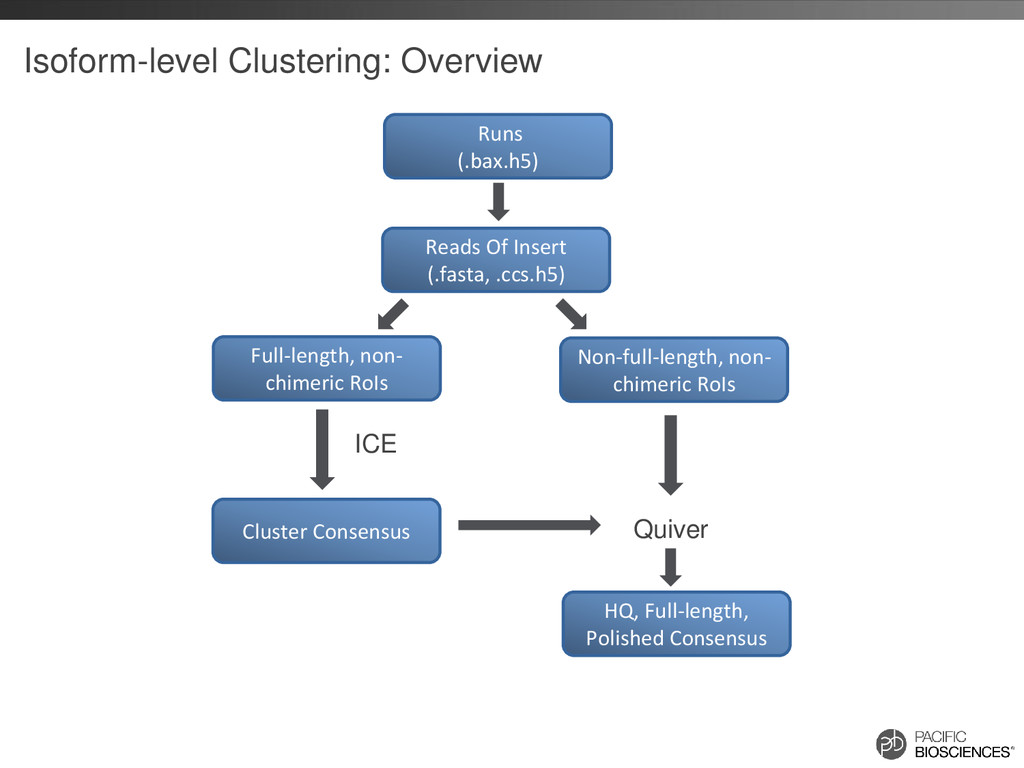
– one read = one transcript • Sequence transcript in full length – most transcripts 1 – 5 kb – PacBio’s avg. read length ~ 5 kb – no assembly required • No systematic bias – GC-rich, AT-rich, tandem repeats
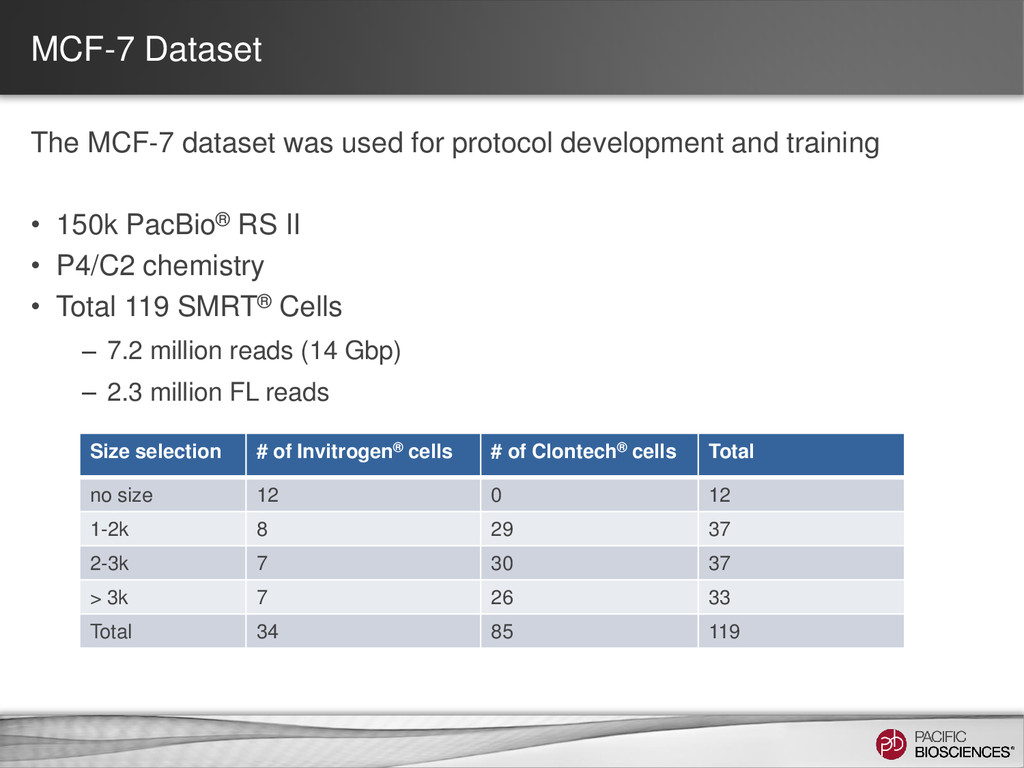
Primer-ligated cDNA form concatemers High incidence of artificial chimera (identifiable cDNA primer in the middle) MCF=7 Clontech 1 – 2 kb Trainee Artificial chimeras A 2415 (3.9%) B 79 (0.5%) C 304 (0.2%) D 235 (0.2%) (AAA)n Artificial Concatemer 5’ primer Transcript 1 Transcript 2 3’ primer 3’ primer 5’ primer
differs depending on transcript size range • Detect and Remove Artificial Chimeras – Artificial concatemers are rare (~0.2%) and avoidable by increasing SMRT® adapter concentration – PCR chimeras are difficult to completely avoid (~3%) but can be detected computationally (if reference genome available), however there are also biological chimeras 25
(top, red) capture multiple isoforms of the BRCA1 gene. Additionally the nearby NBR2 transcript, which is thought to be a non-coding gene that shares a bi-directional promoter with BRCA1, is also observed.
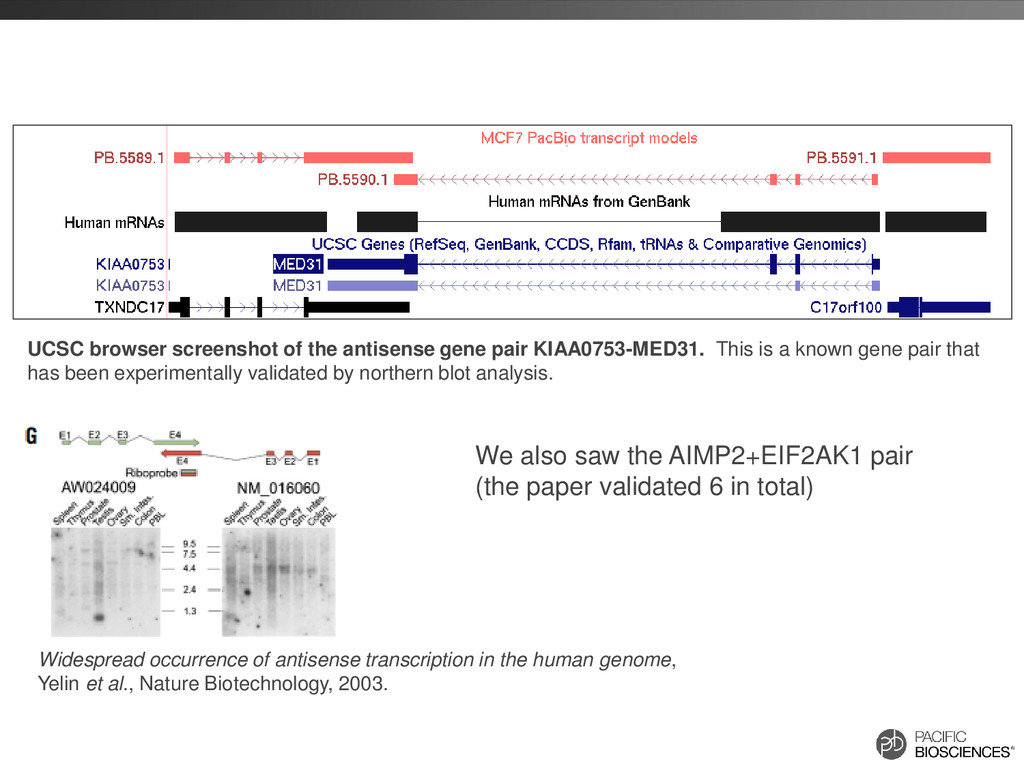
is a known gene pair that has been experimentally validated by northern blot analysis. Widespread occurrence of antisense transcription in the human genome, Yelin et al., Nature Biotechnology, 2003. We also saw the AIMP2+EIF2AK1 pair (the paper validated 6 in total)
distinct coding loci • Use genomic aligners (GMAP) to find fusion candidates • However, PCR chimeras can form during library preparation and are hard to distinguish from true cancer fusion genes • Current solution: create several “filtering steps” – require a minimum number of full-length, raw-read support – require that each mapped locus encodes a different gene • Post-filtering: 93 fusion candidates
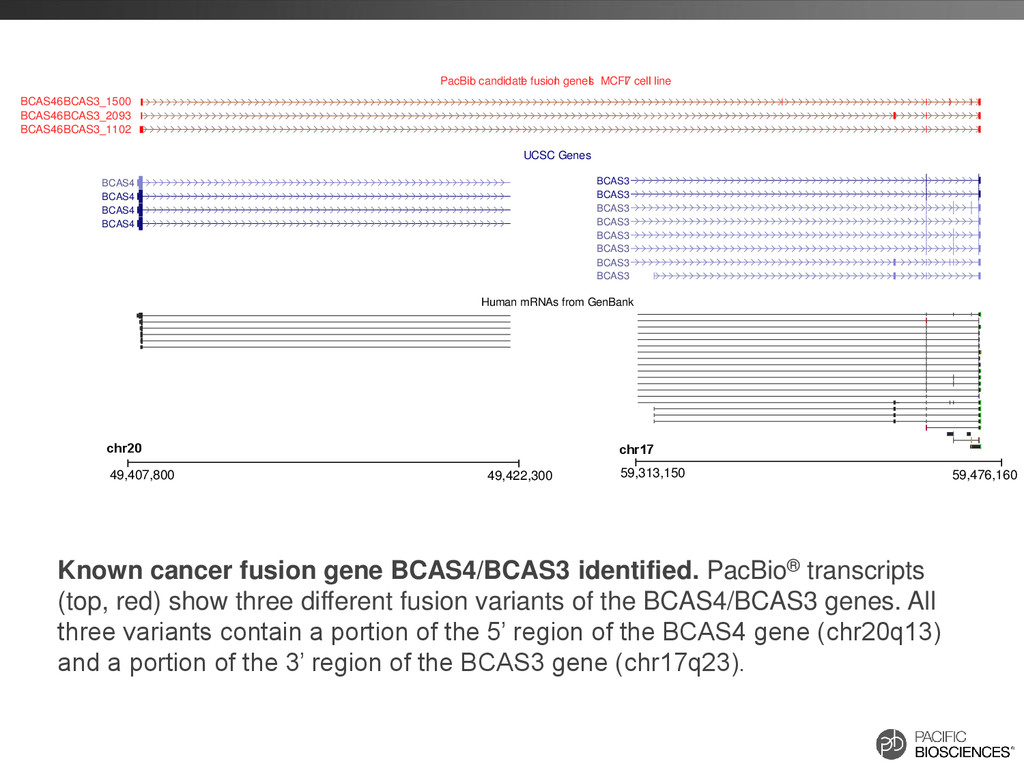
BCAS4 BCAS4 BCAS46BCAS3_1500 BCAS46BCAS3_2093 BCAS46BCAS3_1102 PacBio l candidate l fusion l genes l l l - l l l MCF7 l cell l line UCSC Genes Human mRNAs from GenBank chr20 49,407,800 49,422,300 chr17 59,313,150 59,476,160 Known cancer fusion gene BCAS4/BCAS3 identified. PacBio® transcripts (top, red) show three different fusion variants of the BCAS4/BCAS3 genes. All three variants contain a portion of the 5’ region of the BCAS4 gene (chr20q13) and a portion of the 3’ region of the BCAS3 gene (chr17q23).
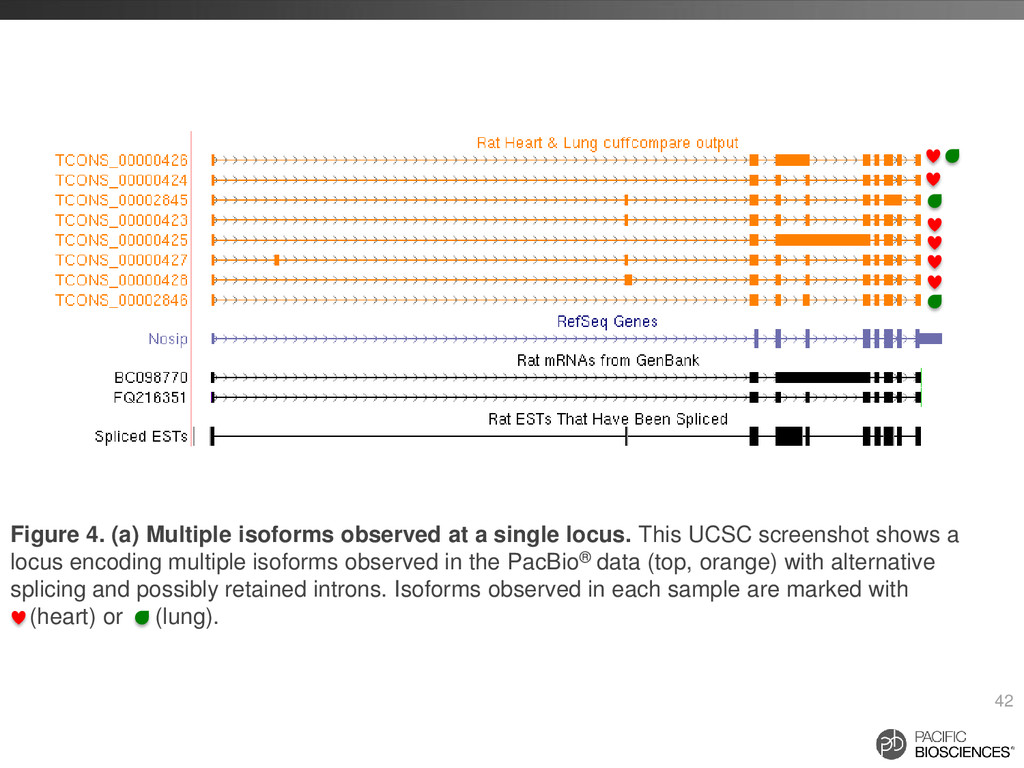
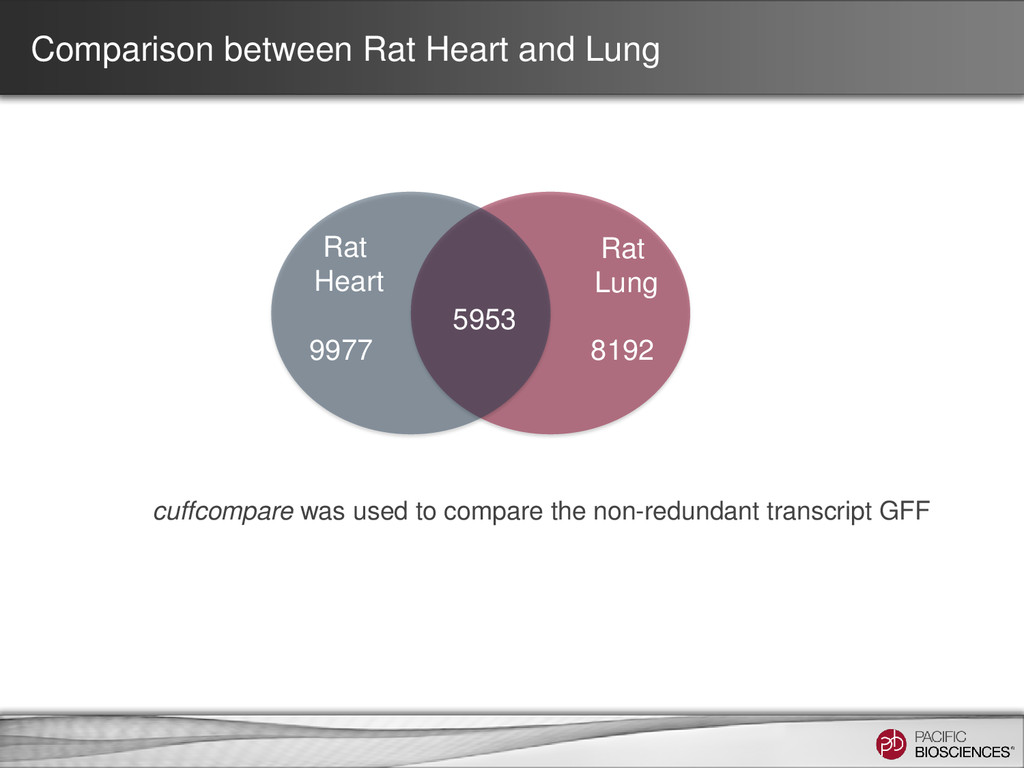
locus. This UCSC screenshot shows a locus encoding multiple isoforms observed in the PacBio® data (top, orange) with alternative splicing and possibly retained introns. Isoforms observed in each sample are marked with (heart) or (lung).
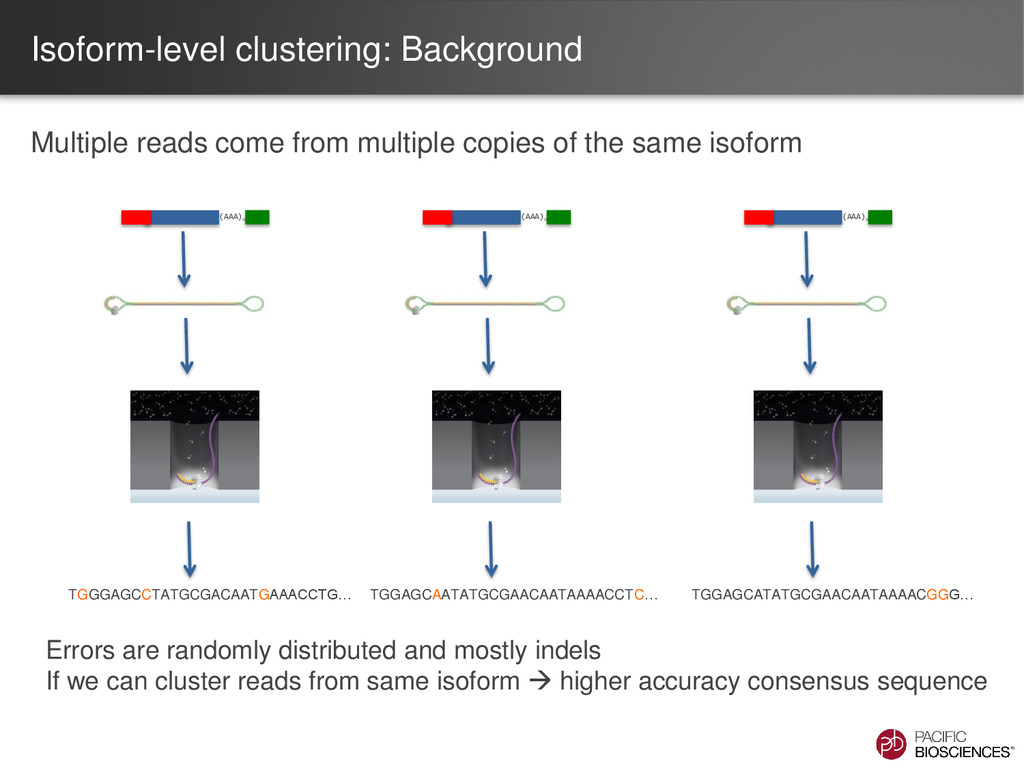
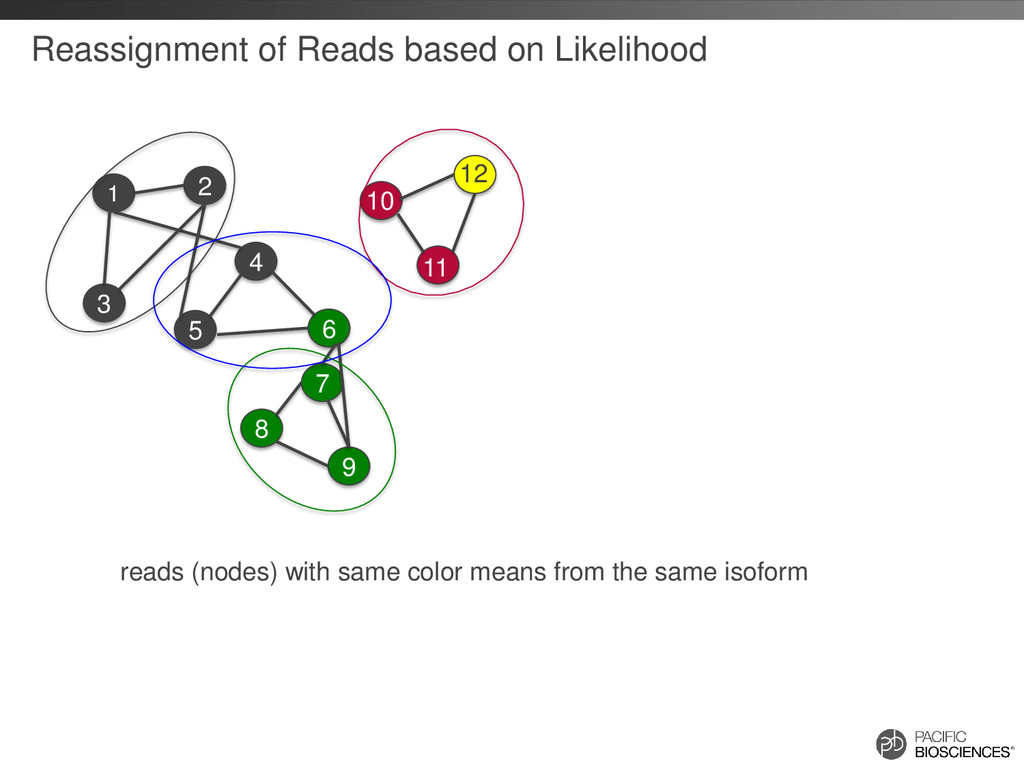
the same isoform (AAA)n TGGGAGCCTATGCGACAATGAAACCTG… (AAA)n TGGAGCAATATGCGAACAATAAAACCTC… (AAA)n TGGAGCATATGCGAACAATAAAACGGG… Errors are randomly distributed and mostly indels If we can cluster reads from same isoform higher accuracy consensus sequence
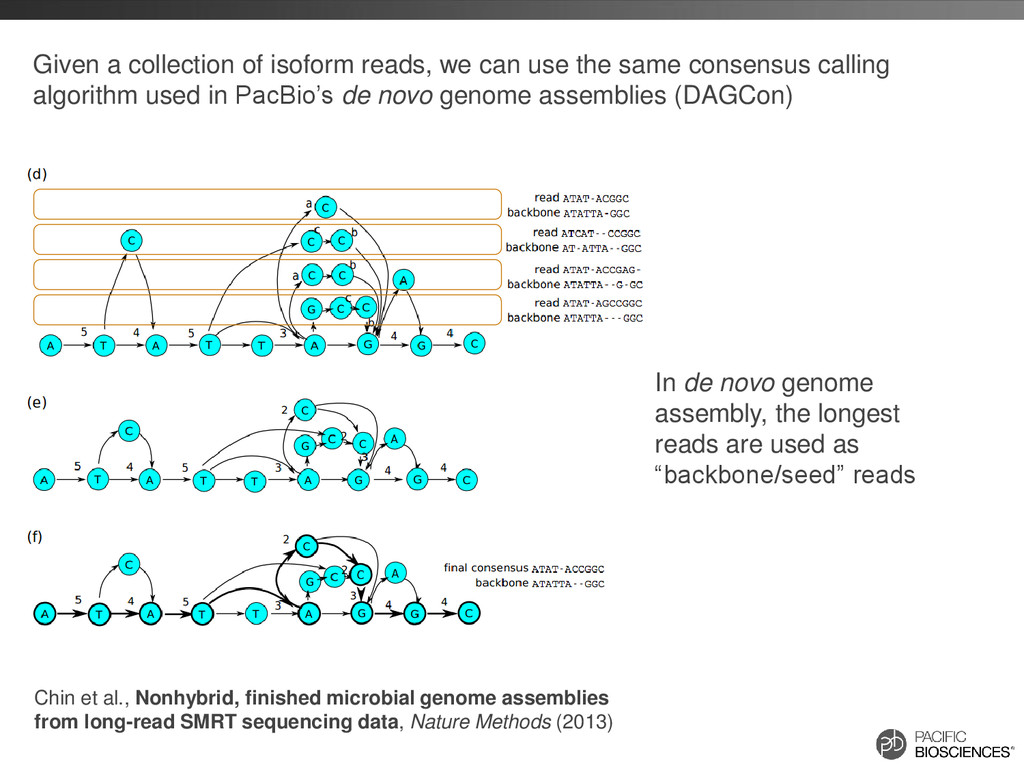
same consensus calling algorithm used in PacBio’s de novo genome assemblies (DAGCon) Chin et al., Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data, Nature Methods (2013) In de novo genome assembly, the longest reads are used as “backbone/seed” reads
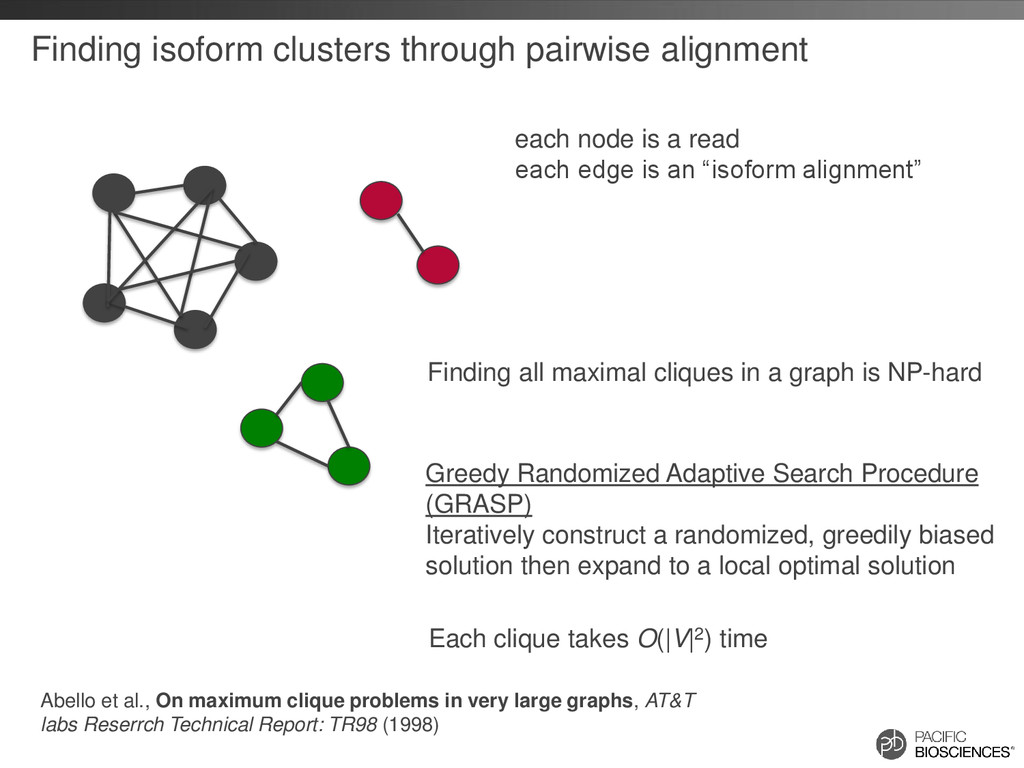
read each edge is an “isoform alignment” Finding all maximal cliques in a graph is NP-hard Abello et al., On maximum clique problems in very large graphs, AT&T labs Reserrch Technical Report: TR98 (1998) Greedy Randomized Adaptive Search Procedure (GRASP) Iteratively construct a randomized, greedily biased solution then expand to a local optimal solution Each clique takes O(|V|2) time
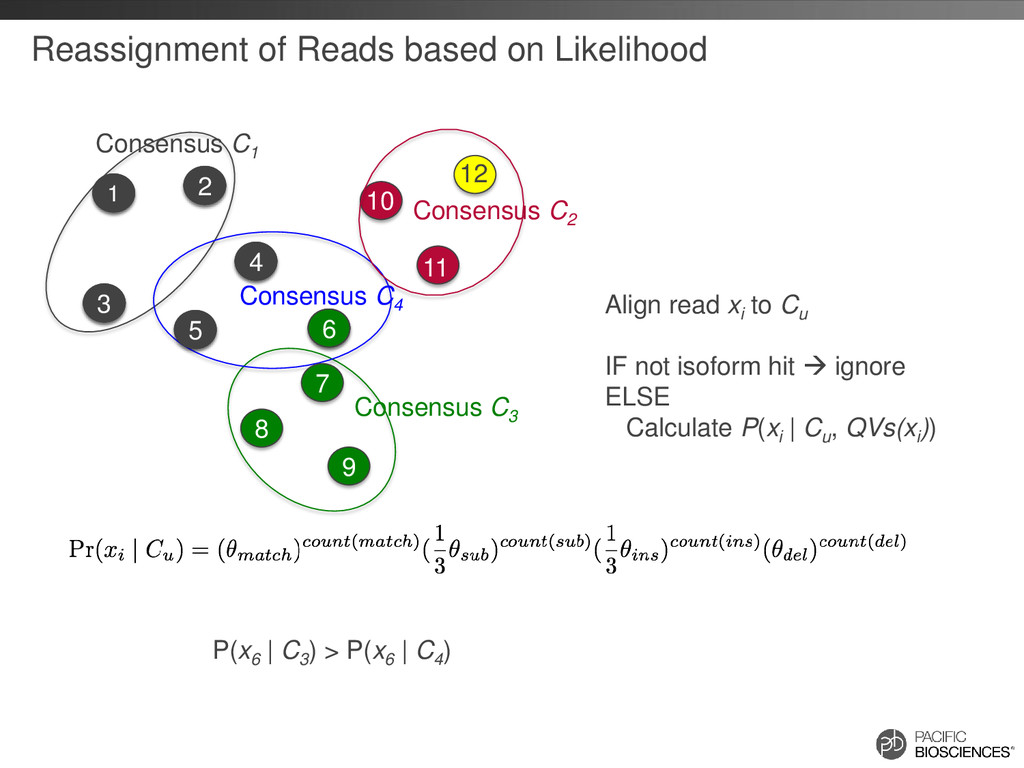
+ D + 745 GAACAGT--CAGCGTATTGTCAGGGGCAGAAATAGTGAAGGAC-------AGAAAAA |||||||**|||||||||||||||||||||||||||**||*||*******||||||| 746 GAACAGTCACAGCGTATTGTCAGGGGCAGAAATAGT--AGTACCAAAAAAAGAAAAA S + I ++++++ D + 000000011000000000000000000000000000110010000000010000000 Difference Array Every base has QV for: • substitution • insertion • deletion Look for region [i, j] where j – i ≥ T and sum(D[i:j]) ≥ C * T C = 0.5, T = 10 If no such region can be found, then consider the two reads to be from the same isoform Tseng & Tompa, Algorithms for locating extremely conserved elements in multiple sequence alignments, BMC Bioinformatics (2009)
+ D + 745 GAACAGT--CAGCGTATTGTCAGGGGCAGAAATAGTGAAGGAC-------AGAAAAA |||||||**|||||||||||||||||||||||||||**||*||*******||||||| 746 GAACAGTCACAGCGTATTGTCAGGGGCAGAAATAGT--AGTACCAAAAAAAGAAAAA S + I D + 000000011000000000000000000000000000110010011111110000000 Difference Array Every base has QV for: • substitution • insertion • deletion Look for region [i, j] where j – i ≥ T and sum(D[i:j]) ≥ C * T C = 0.5, T = 10 If such region is found, then consider two reads as from different isoforms Tseng & Tompa, Algorithms for locating extremely conserved elements in multiple sequence alignments, BMC Bioinformatics (2009)
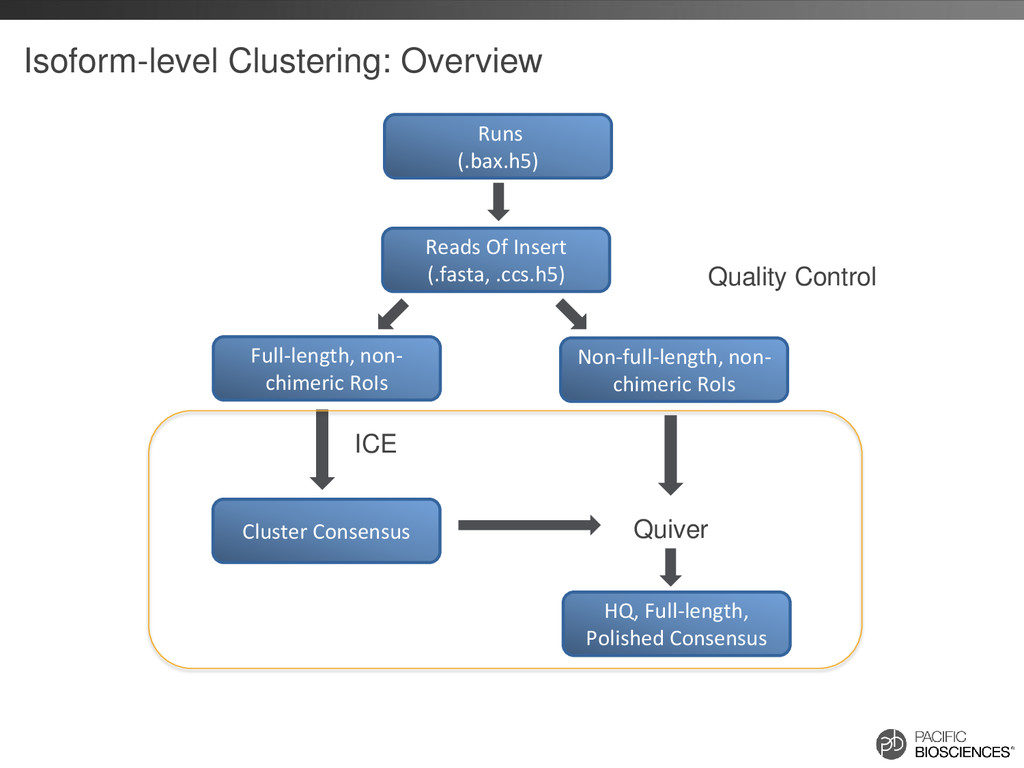
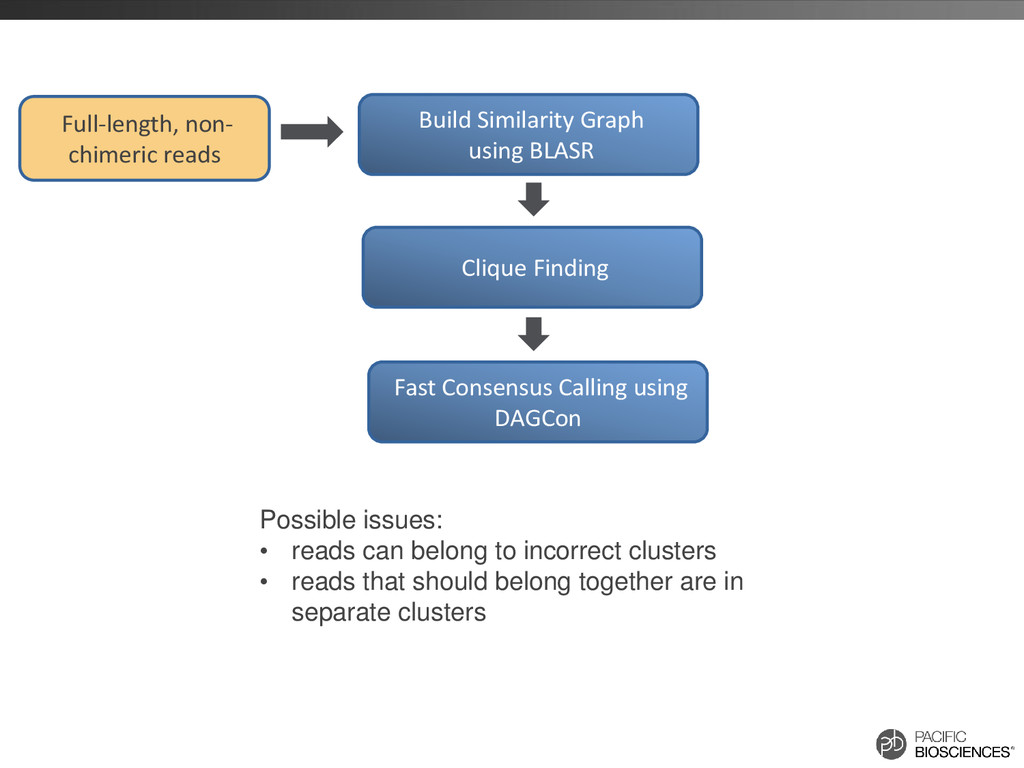
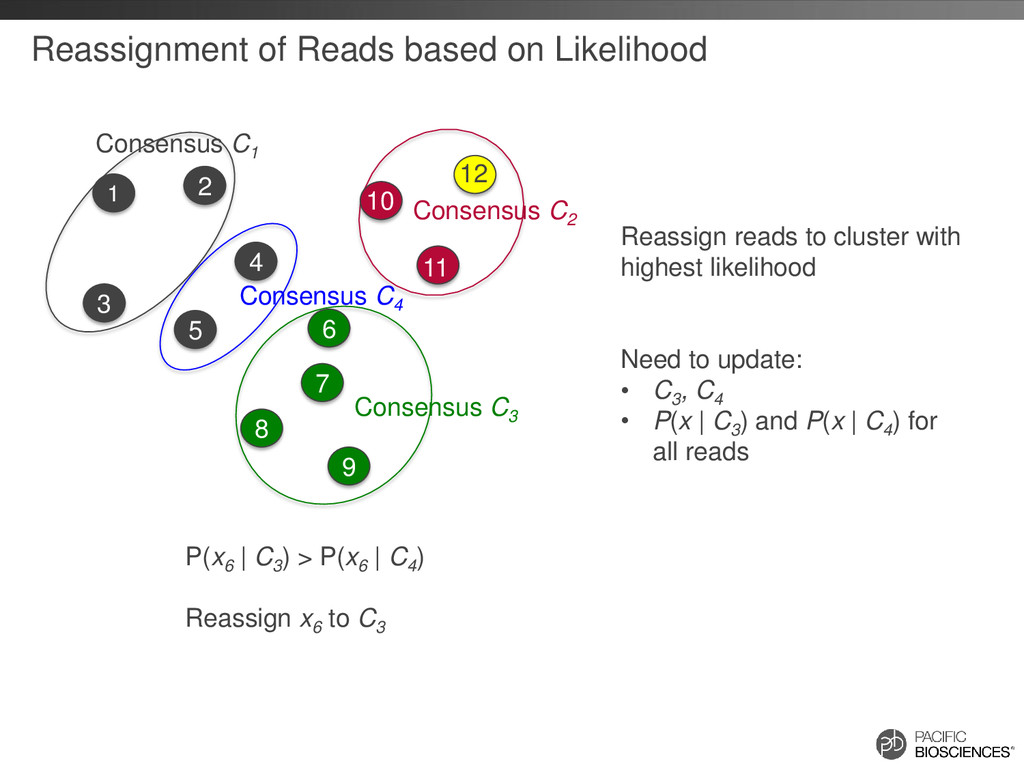
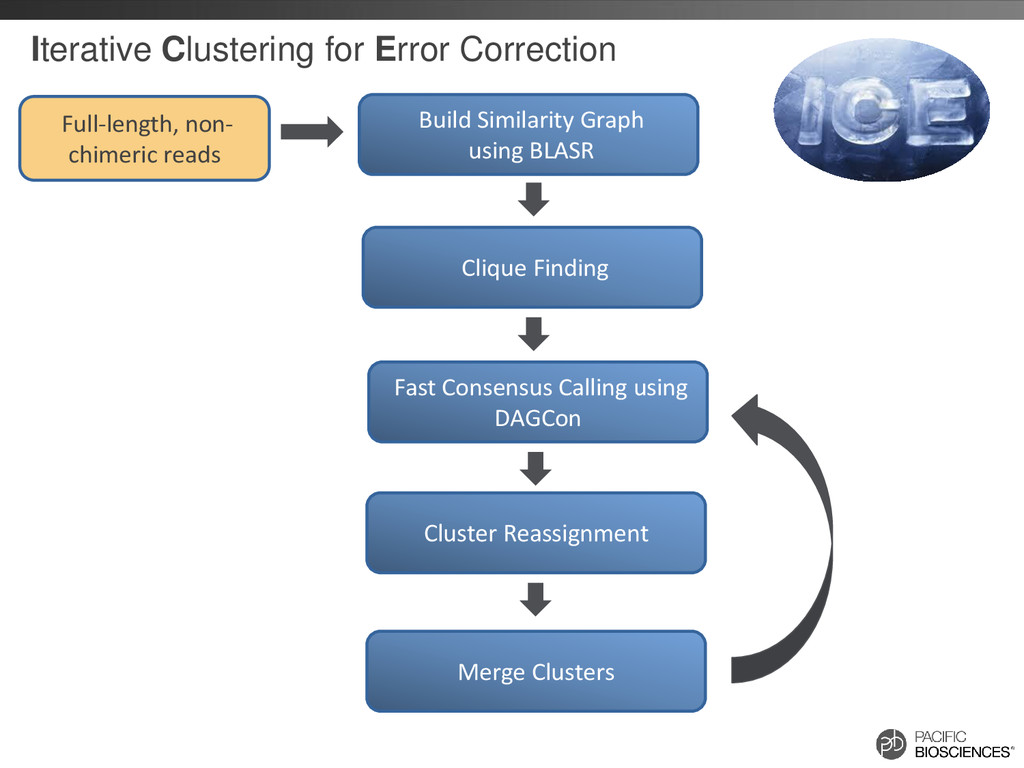
reads that should belong together are in separate clusters Build Similarity Graph using BLASR Clique Finding Fast Consensus Calling using DAGCon Full-length, non- chimeric reads
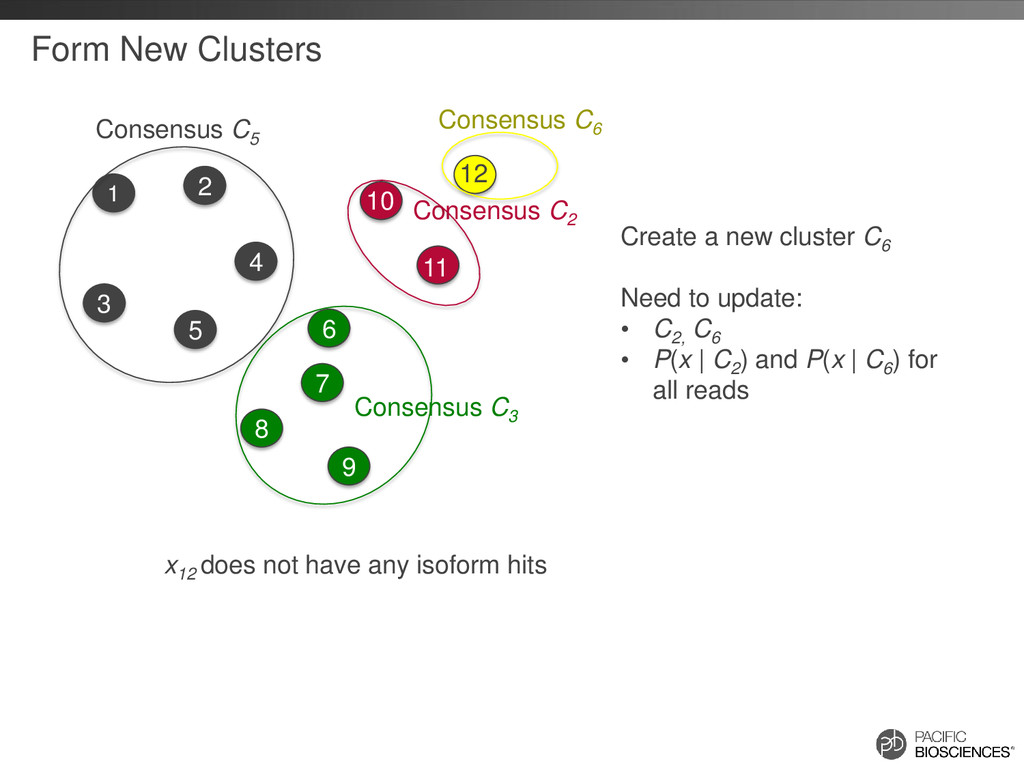
2 5 6 7 8 9 10 11 12 x12 does not have any isoform hits Consensus C5 Create a new cluster C6 Need to update: • C2, C6 • P(x | C2 ) and P(x | C6 ) for all reads Consensus C6
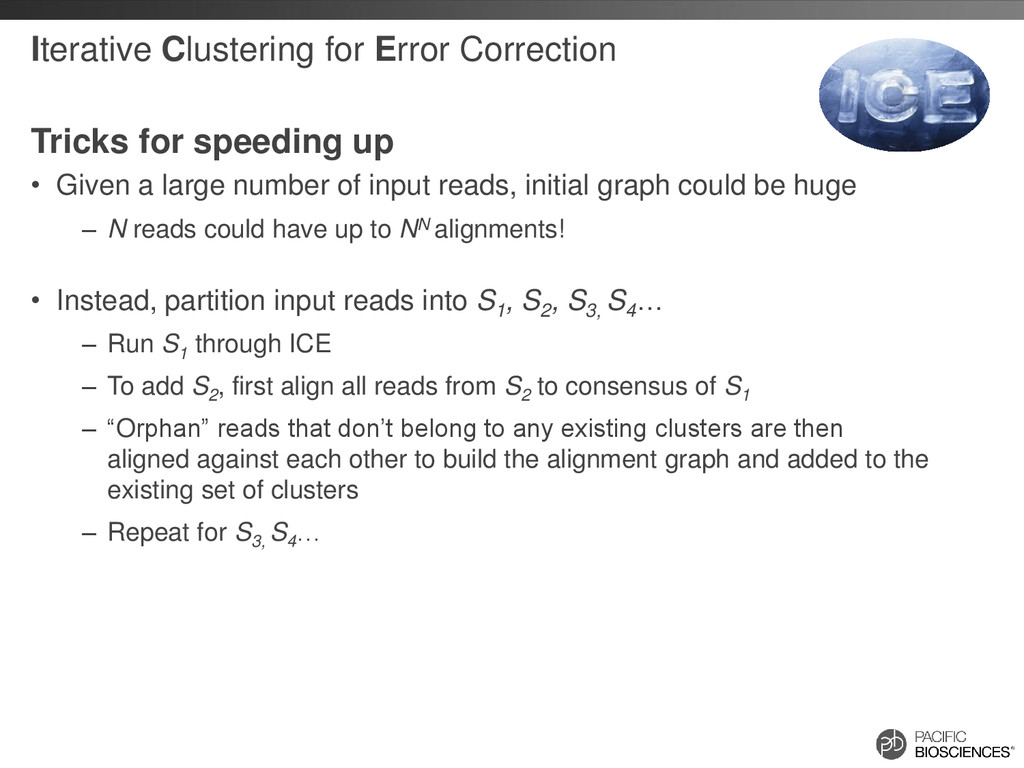
Given a large number of input reads, initial graph could be huge – N reads could have up to NN alignments! • Instead, partition input reads into S1 , S2 , S3, S4 … – Run S1 through ICE – To add S2 , first align all reads from S2 to consensus of S1 – “Orphan” reads that don’t belong to any existing clusters are then aligned against each other to build the alignment graph and added to the existing set of clusters – Repeat for S3, S4 …
Same “isoform hit” criteria – But does not require each read to be fully aligned – Each non-FL read can belong to multiple clusters Chin et al., Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data, Nature Methods (2013)
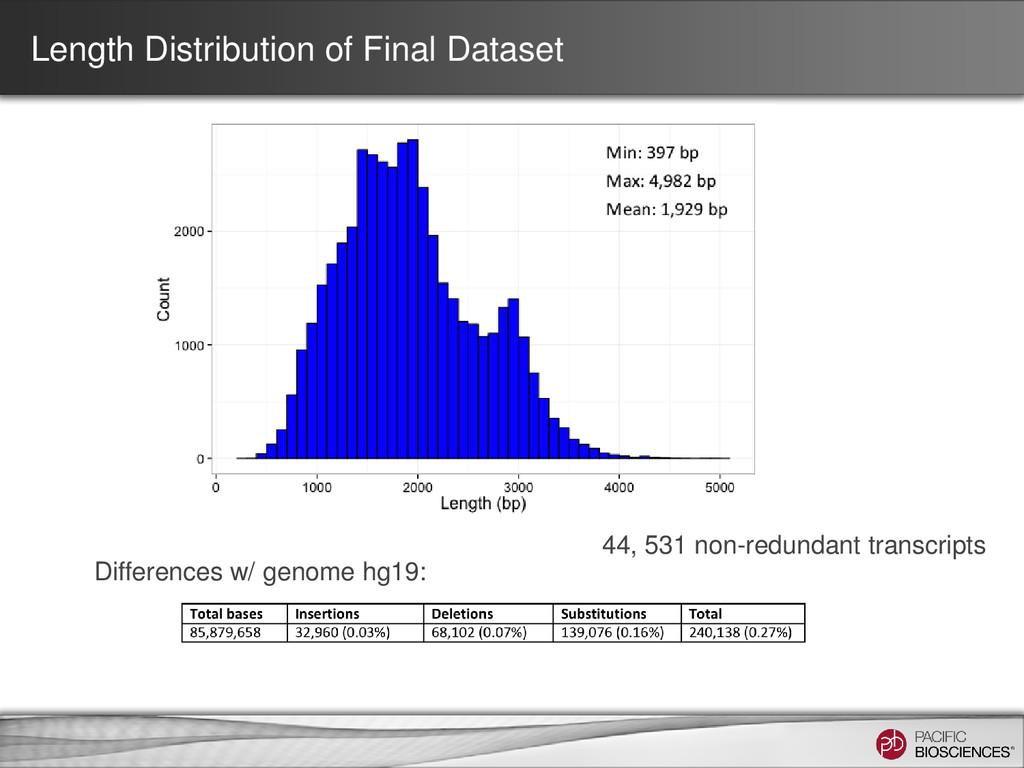
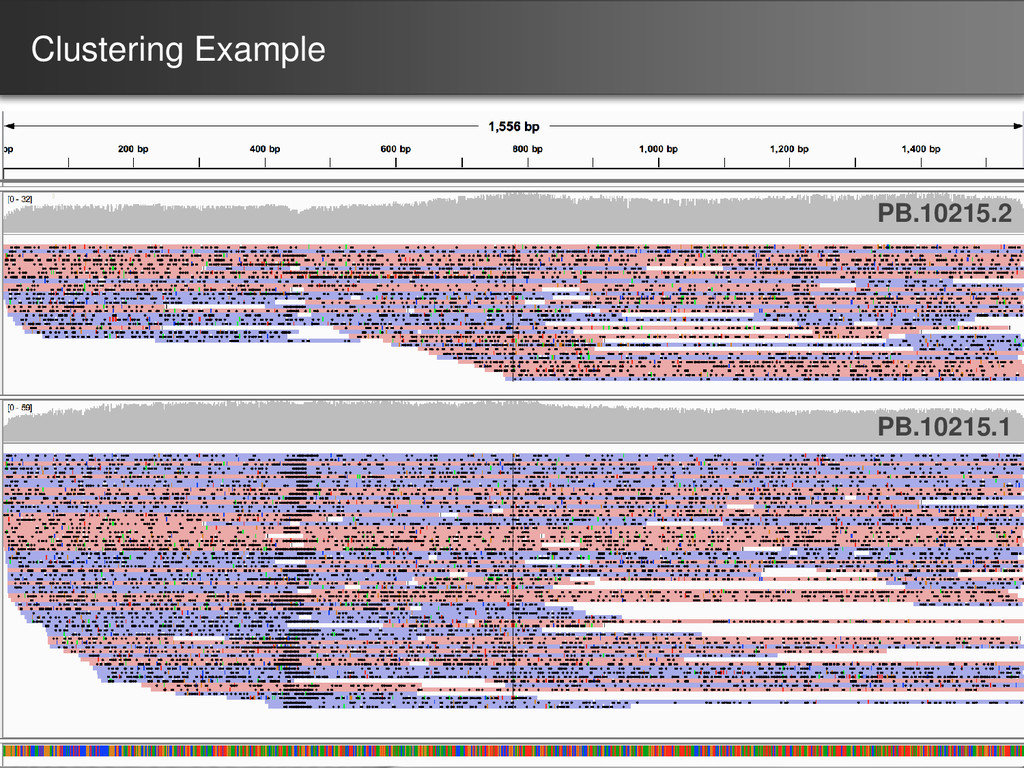
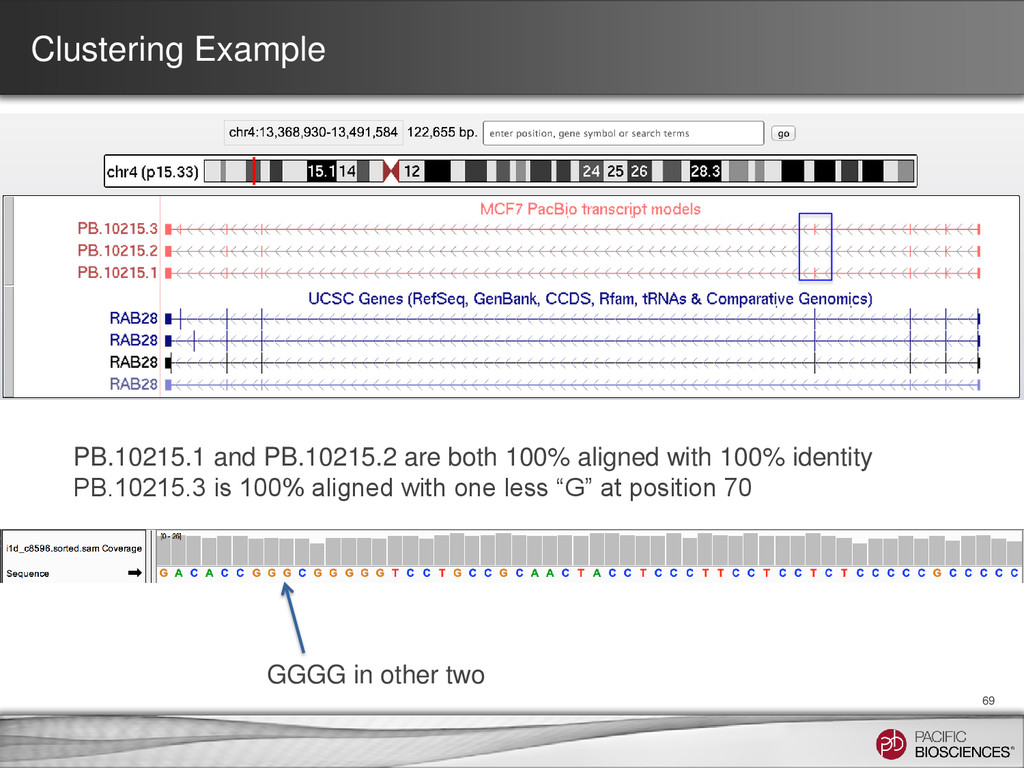
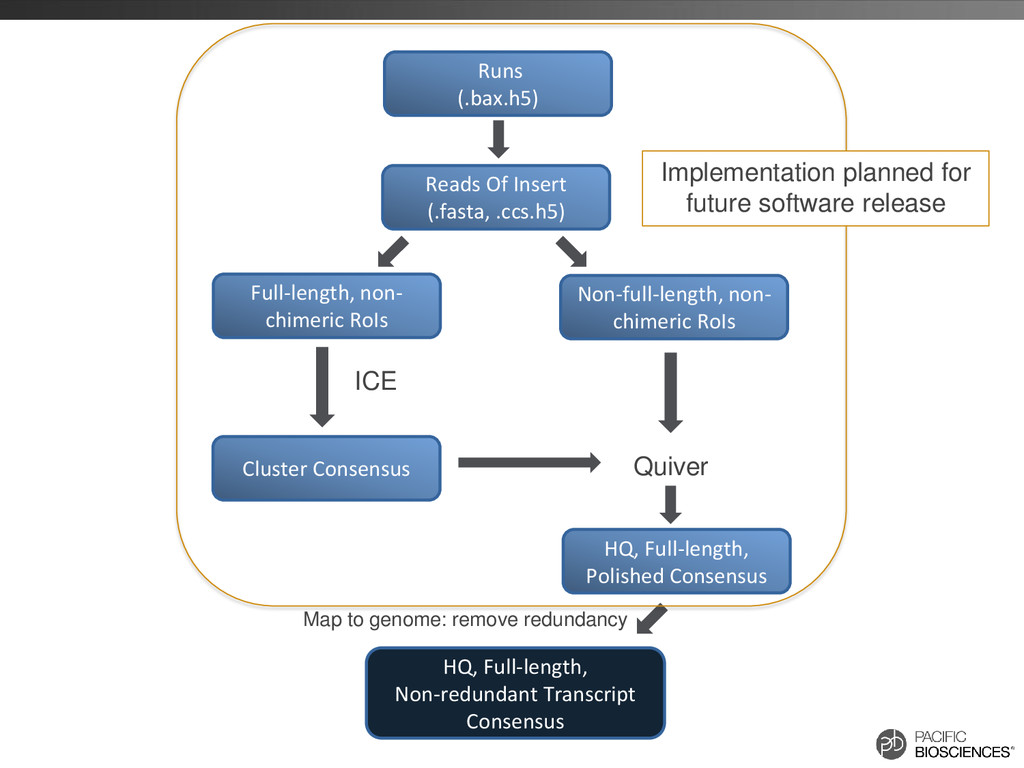
were further processed for collapsing redundant transcripts • Consensus transcripts were mapped back to the genome – If exon structure identical but only differ on the 5’ start site, collapse 70
(GitHub) Code Repository and Tutorial Wiki • Iso-Seq™ Library Preparation Protocol Recent Customer Publications: • Sharon et al., A single-molecule long-read survey of the human transcriptome, Nature Biotech. (2013) • Au et al., Characterization of the human ESC transcriptome by hybrid sequencing, PNAS (2013) • Zhang et al., PacBio sequencing of gene families-a case study with wheat gluten genes, Gene (2013) Contact your FAS to learn more!
Iso-Seq are trademarks of Pacific Biosciences in the United States and/or other countries. All other trademarks are the sole property of their respective owners. 75
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}