Full-length Isoform detection using the PacBio® Iso-Seq™ method. After the training, you will be able to • Choose the best protocol for your experimental design. • Understand how the Iso-Seq method works. • Run an Iso_Seq job and understand the reports generated in SMRT® Portal. • SMRT Technology • PacBio System Workflow • General Understanding of SMRT Portal
now is… you take the transcriptome, you blow it up into pieces and then you try to figure out how they all go back together again… If you think about it, it’s kind of a crazy way to do things” Michael Synder Professor and Chair of Genetics Stanford University Tal Nawy, End to end RNA Sequencing, Nature Methods, v10, n10, Dec . 2013, p1144–1145 Ian Korf (2013) Genomics: the state of the art in RNA-seq analysis, Nature Methods, Nov 26;10(12):1165-6. doi: 10.1038/nmeth.2735.
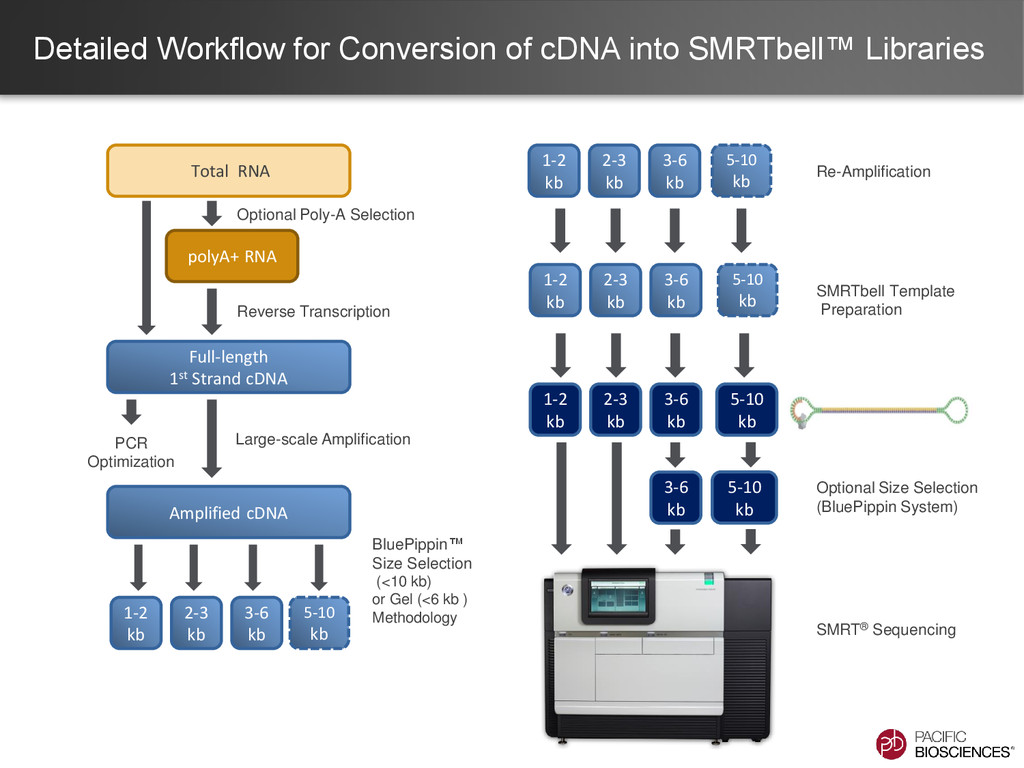
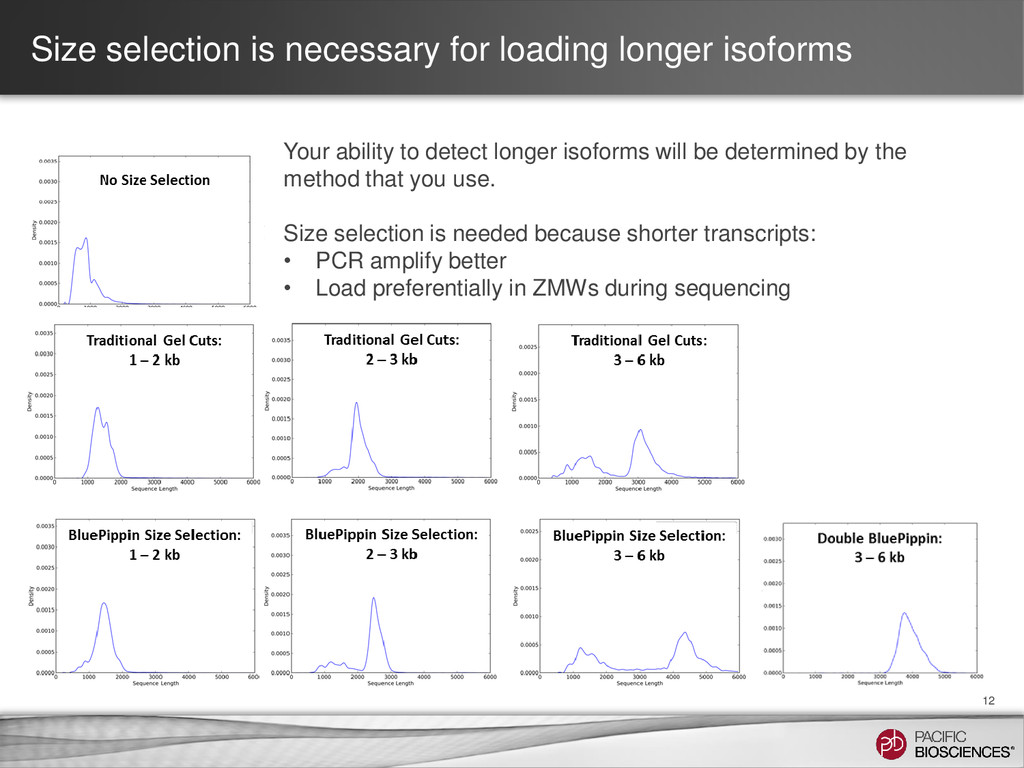
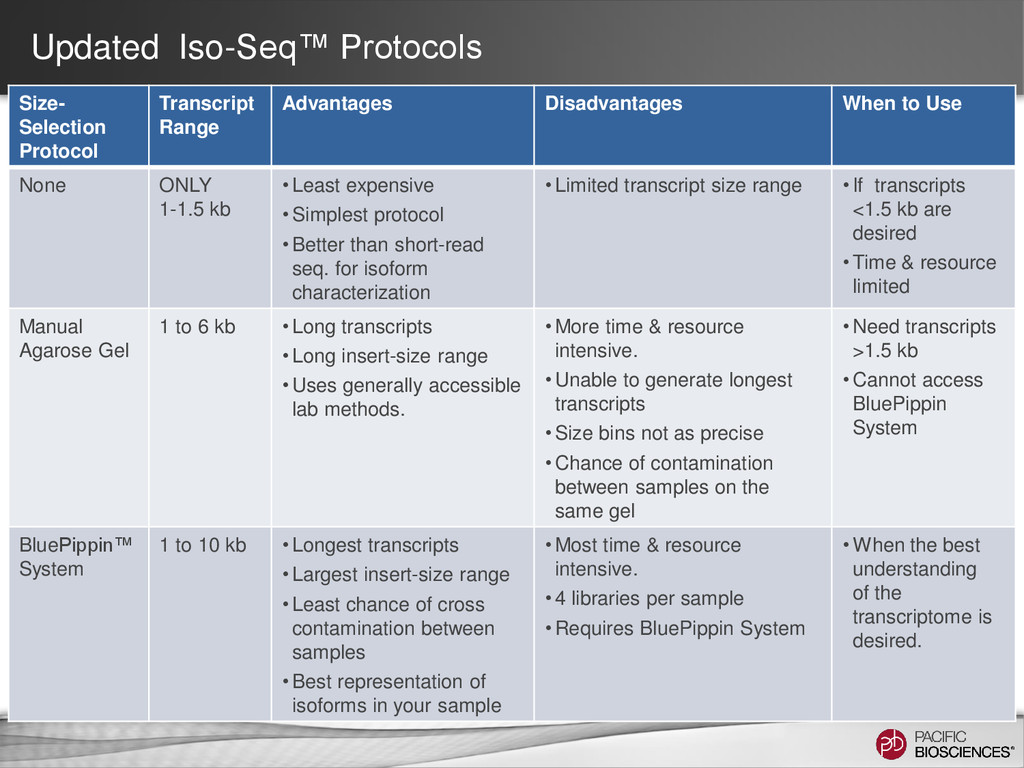
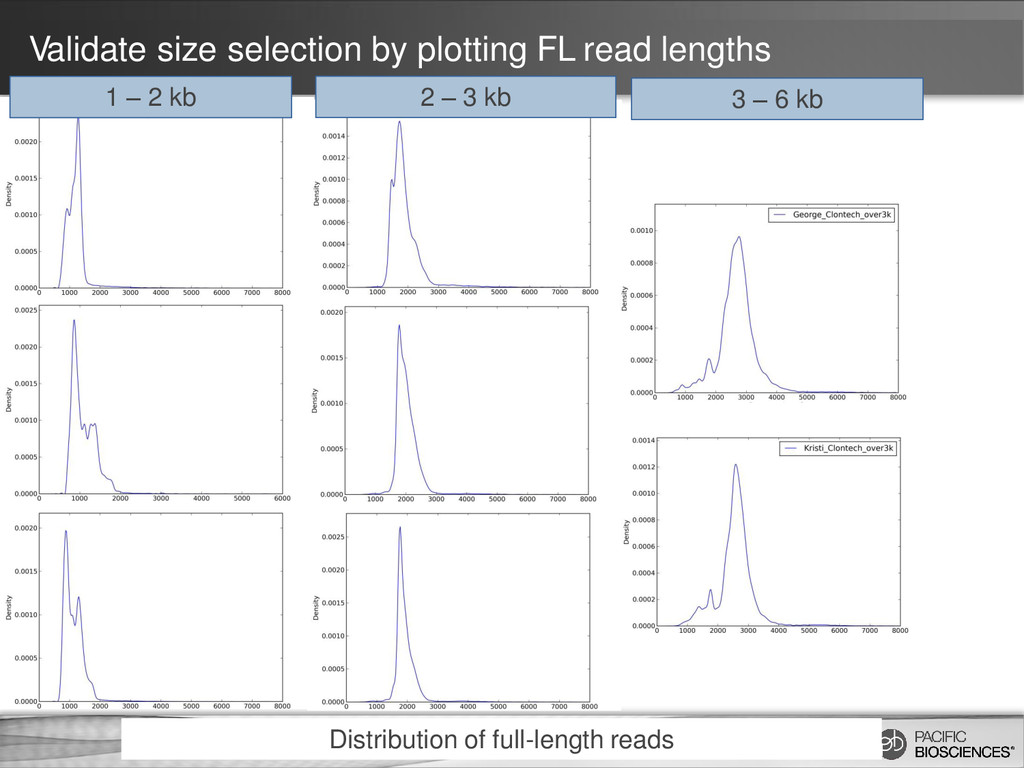
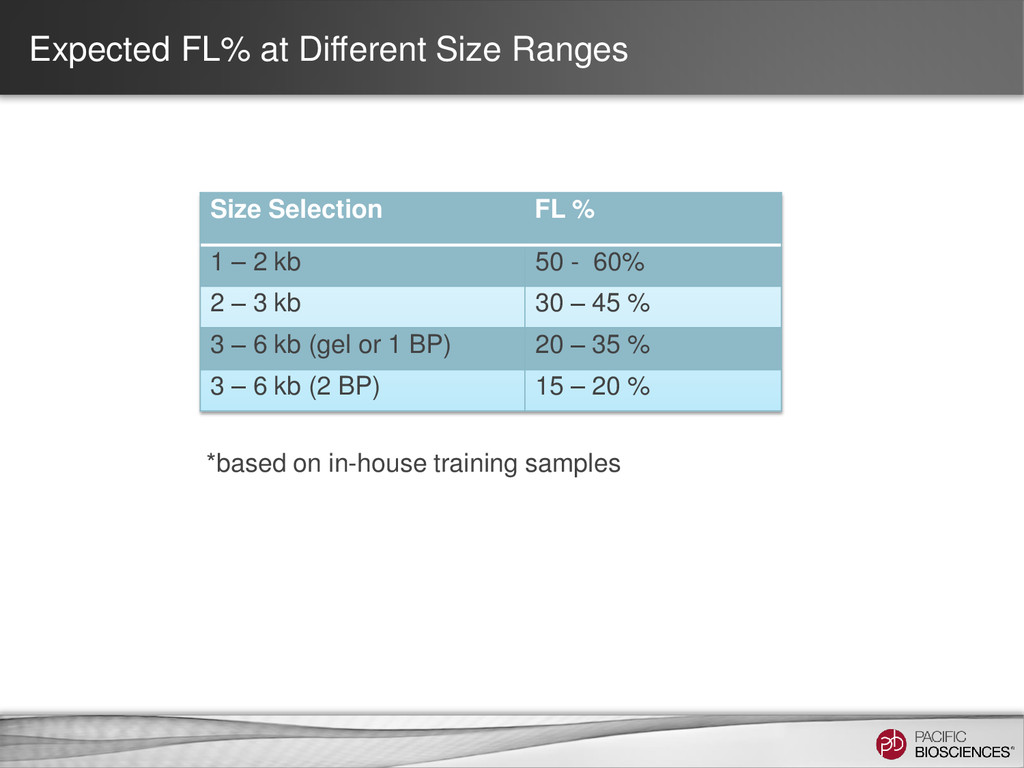
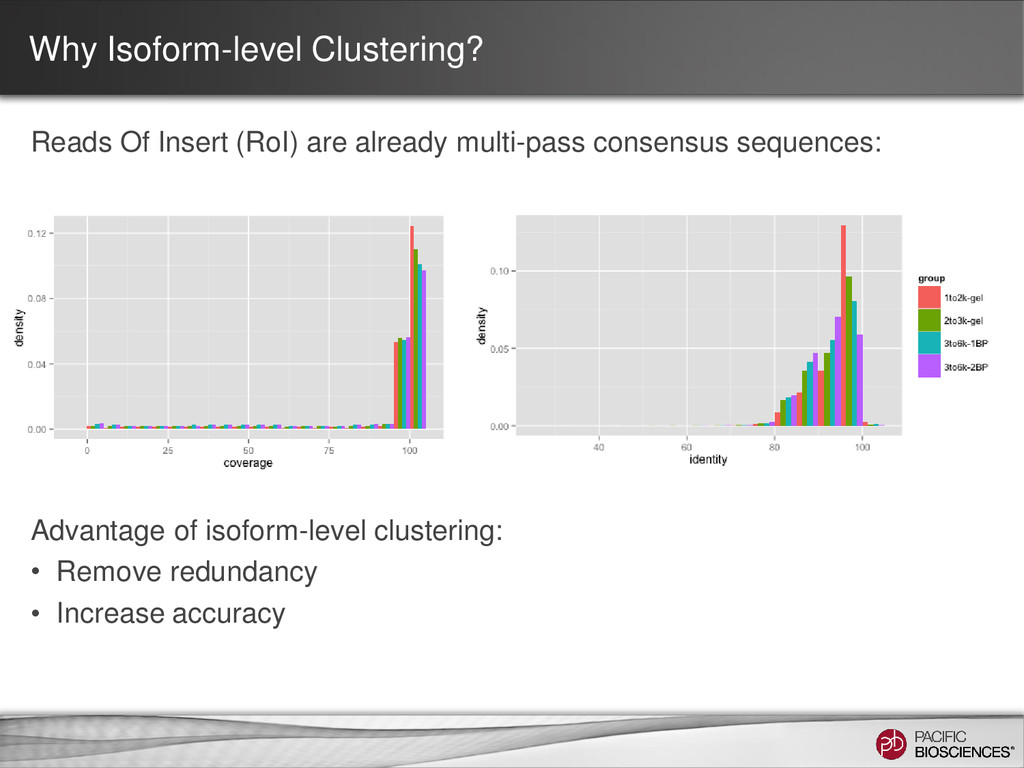
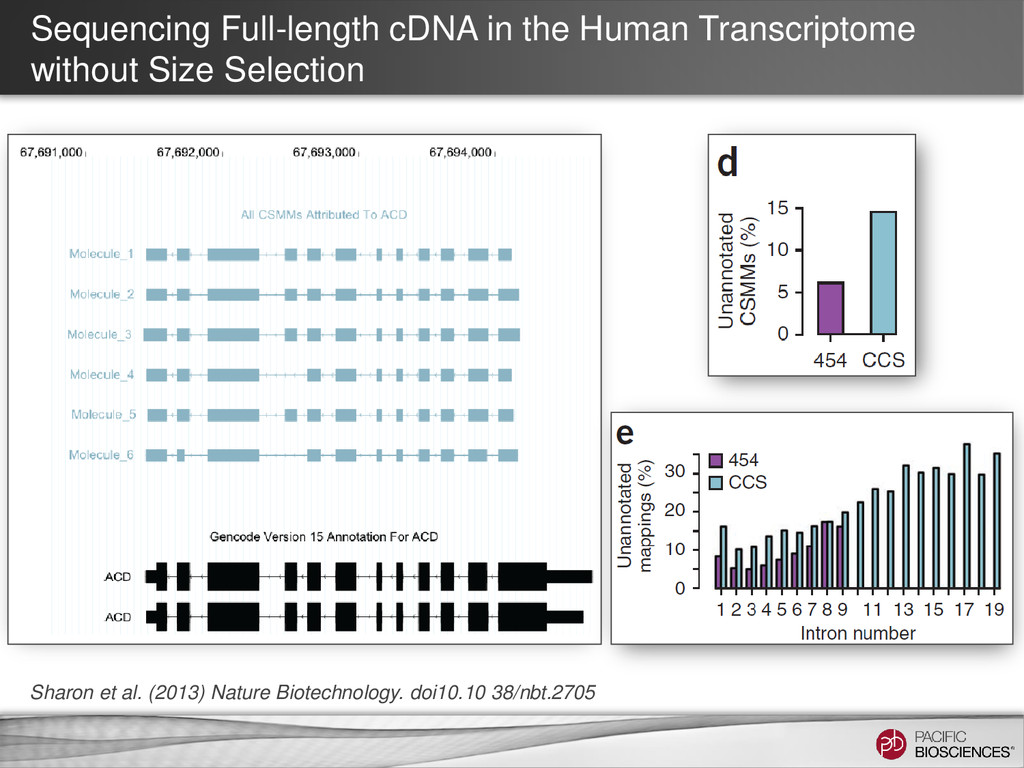
ability to detect longer isoforms will be determined by the method that you use. Size selection is needed because shorter transcripts: • PCR amplify better • Load preferentially in ZMWs during sequencing
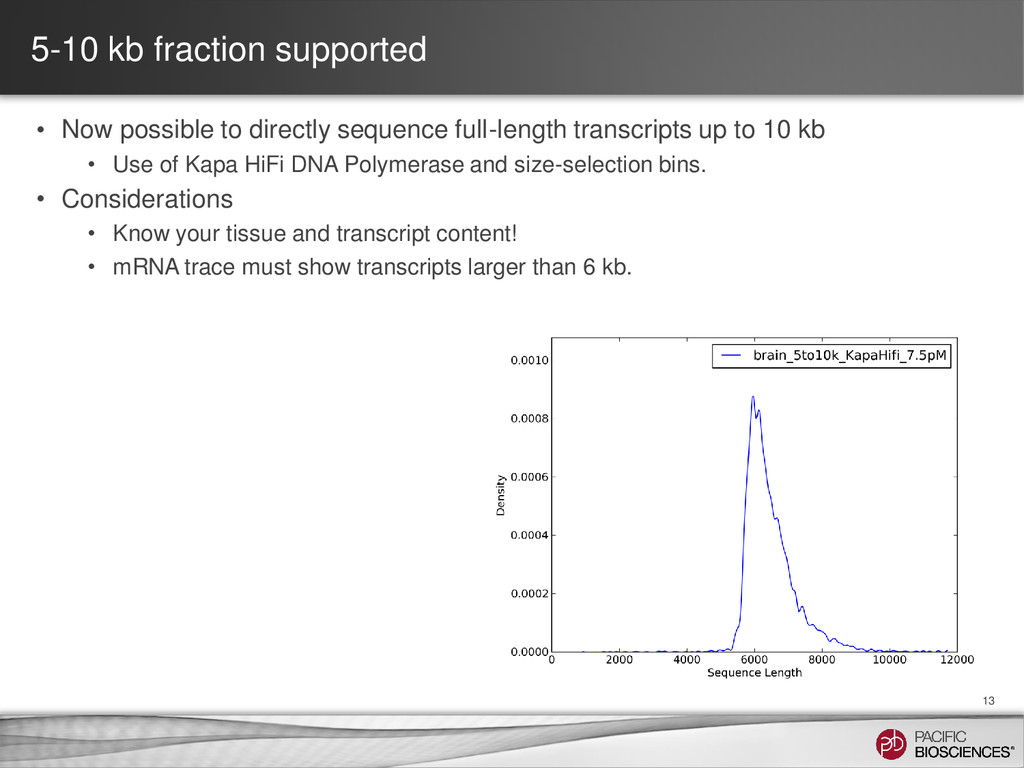
sequence full-length transcripts up to 10 kb • Use of Kapa HiFi DNA Polymerase and size-selection bins. • Considerations • Know your tissue and transcript content! • mRNA trace must show transcripts larger than 6 kb.
When to Use None ONLY 1-1.5 kb • Least expensive • Simplest protocol • Better than short-read seq. for isoform characterization • Limited transcript size range • If transcripts <1.5 kb are desired • Time & resource limited Manual Agarose Gel 1 to 6 kb • Long transcripts • Long insert-size range • Uses generally accessible lab methods. • More time & resource intensive. • Unable to generate longest transcripts • Size bins not as precise • Chance of contamination between samples on the same gel • Need transcripts >1.5 kb • Cannot access BluePippin System BluePippin™ System 1 to 10 kb • Longest transcripts • Largest insert-size range • Least chance of cross contamination between samples • Best representation of isoforms in your sample • Most time & resource intensive. • 4 libraries per sample • Requires BluePippin System • When the best understanding of the transcriptome is desired.
Transcript 2 3’ primer 3’ primer 5’ primer Cause Outcome Detection Low SMRT® adaptor concentration Primer-ligated cDNA form concatemers High incidence of artificial chimera (identifiable cDNA primer in the middle) MCF=7 Clontech 1 – 2 kb Trainee Artificial chimeras A 2415 (3.9%) B 79 (0.5%) C 304 (0.2%) D 235 (0.2%)
< )< n <<<<<<<<<<<<<<<<<<<<<<< 5’ primer Transcript 1, partial, reversed 3’ primer >>>>>>>>>>>>>>>>>>>>>> (AAA)n However, there are also biological chimeras! Cause Outcome Detection PCR amplification Random fusion of ligated transcripts Single read maps to different loci/genes Sample Size Selection Multi-mapped MCF7 1 – 2 kb 2.7% Rat Muscle 1 – 2 kb 3.2% Mouse Liver 1 – 2 kb 2.2% Mouse Liver 2 – 3 kb 1.6%
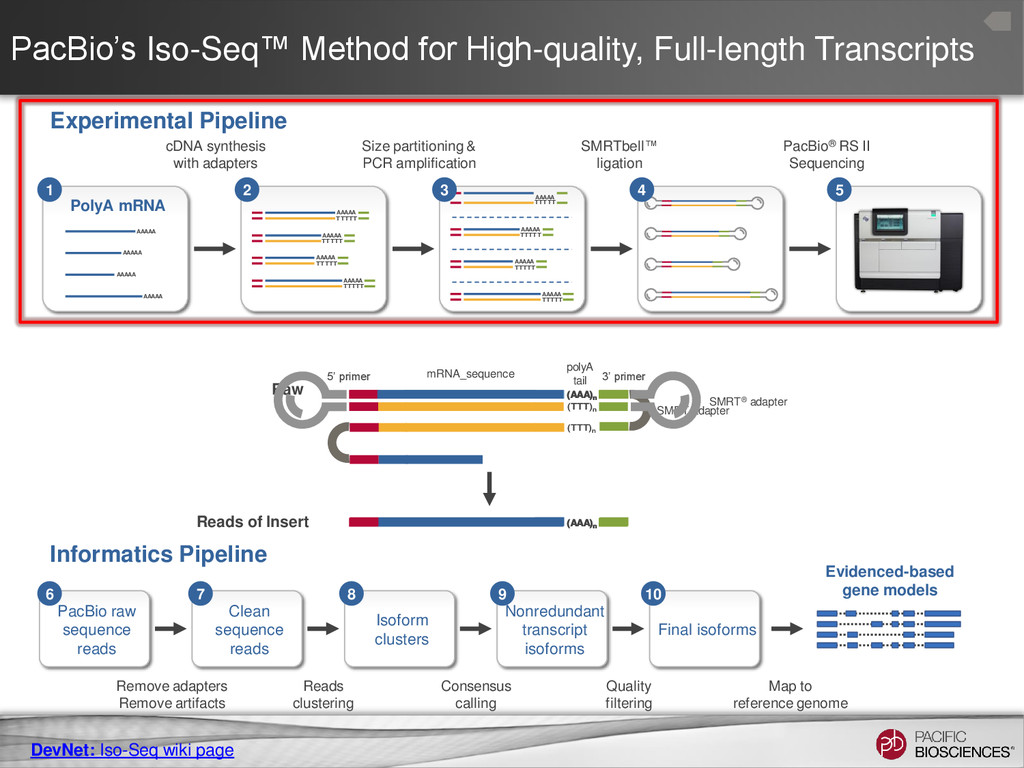
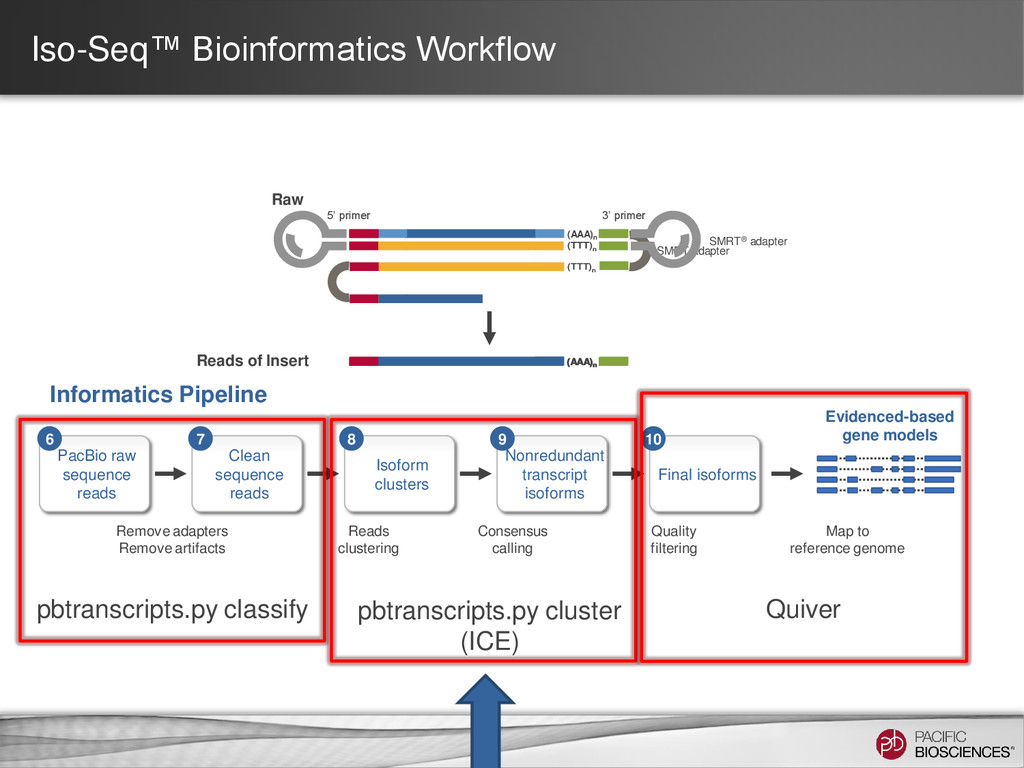
expected full-length reads/cell differs depending on transcript size range. • Detect and Remove Artificial Chimeras – Artificial concatemers are rare (~0.2%) and avoidable by increasing SMRT® adapter concentration. – PCR chimeras are difficult to completely avoid (~3%) but can be detected computationally (if reference genome available). – However, there are also biological chimeras. 25
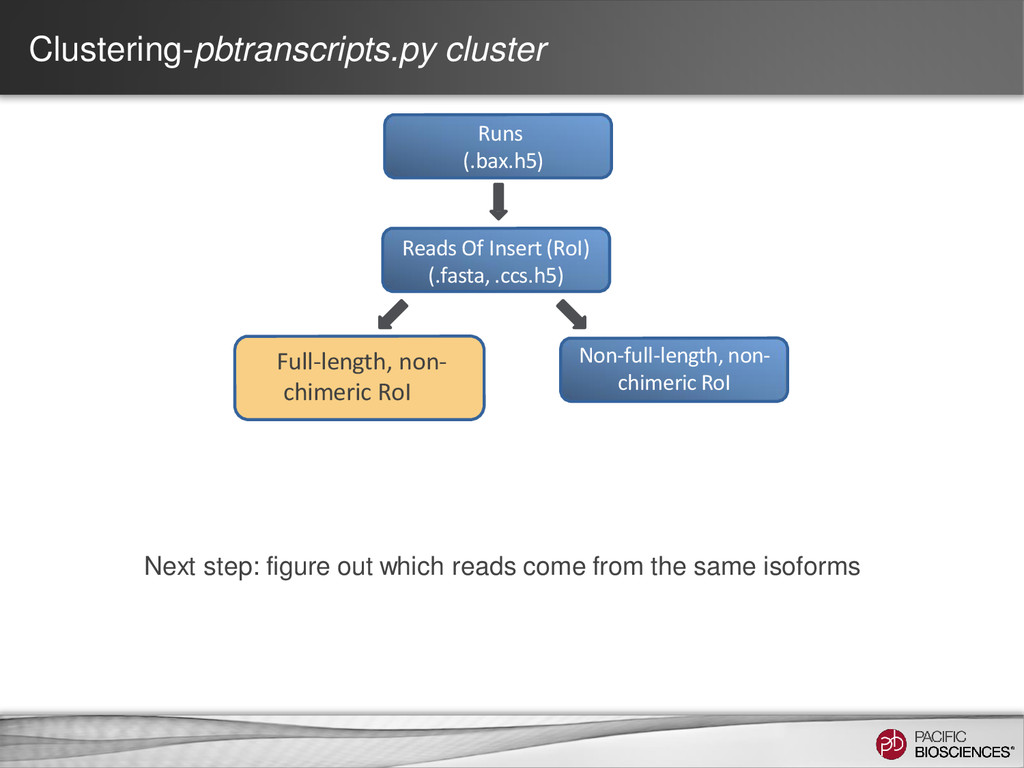
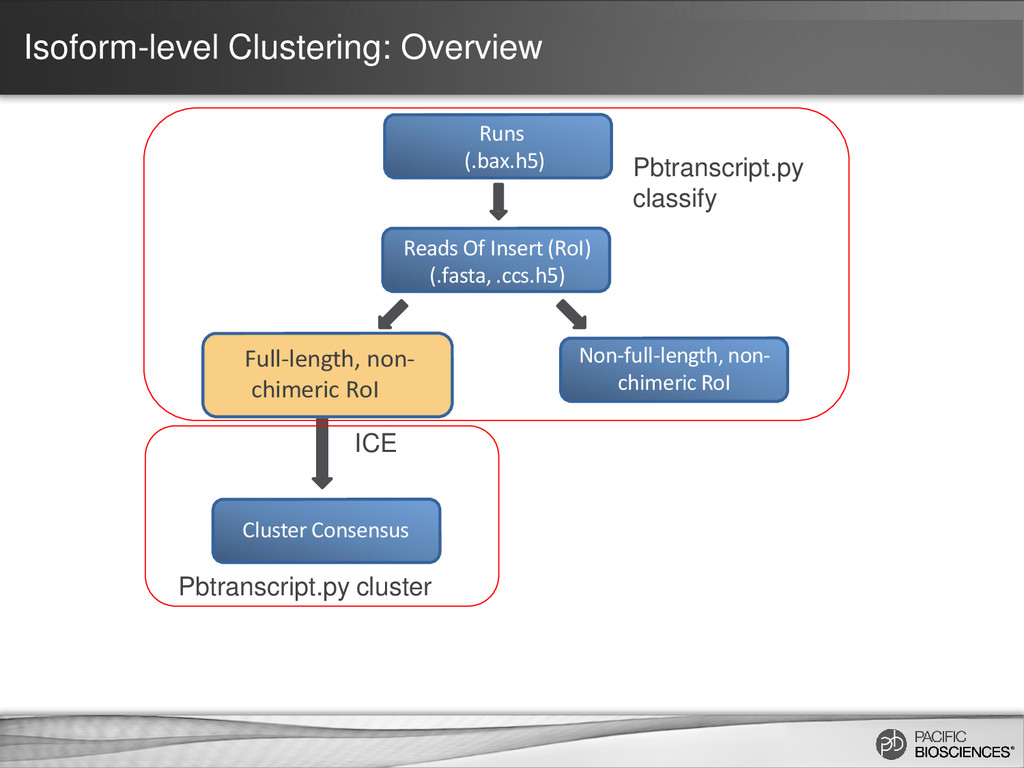
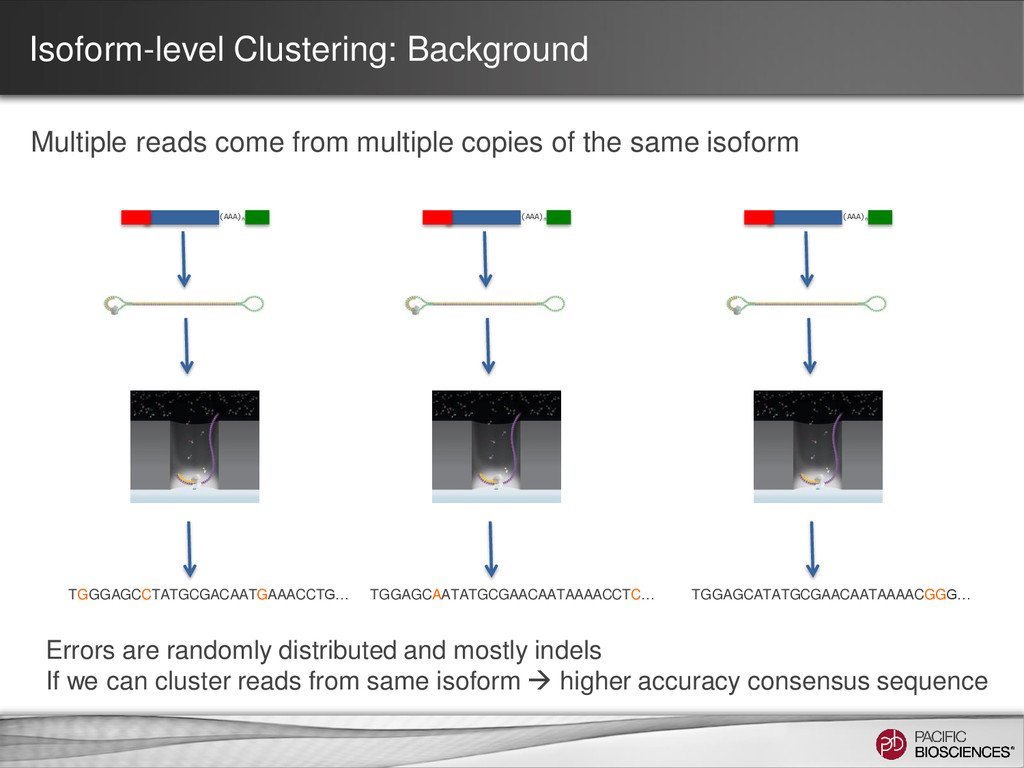
the same isoform (AAA)n TGGGAGCCTATGCGACAATGAAACCTG… (AAA)n TGGAGCAATATGCGAACAATAAAACCTC… (AAA)n TGGAGCATATGCGAACAATAAAACGGG… Errors are randomly distributed and mostly indels If we can cluster reads from same isoform higher accuracy consensus sequence
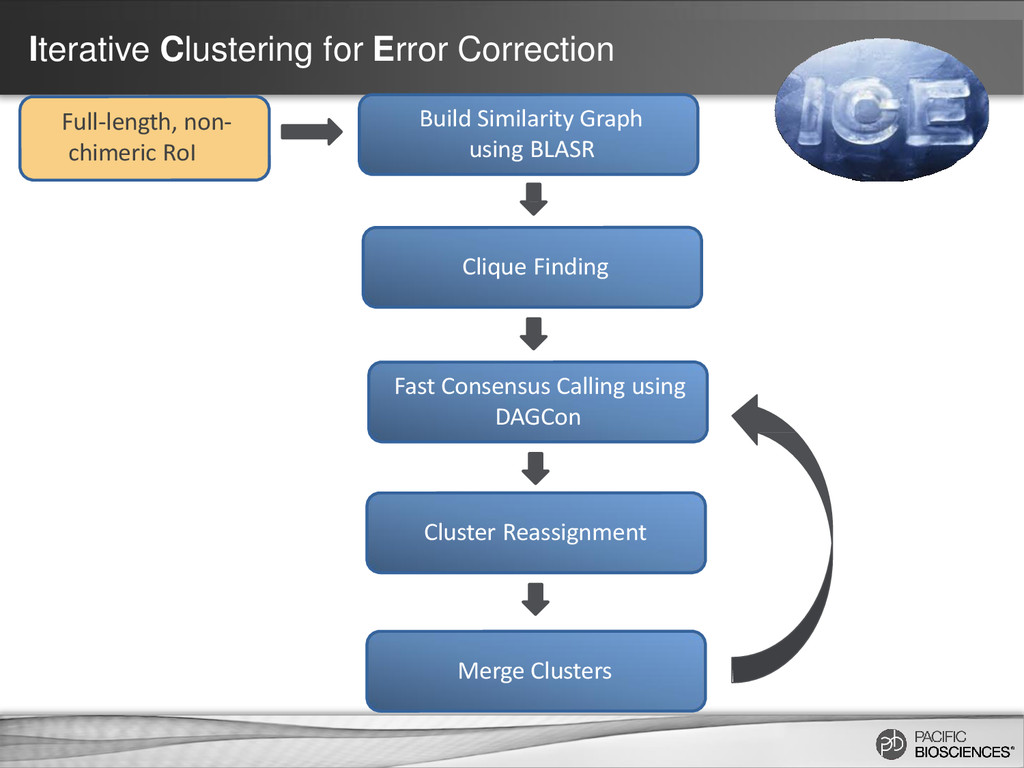
same consensus calling algorithm used in PacBio’s de novo genome assemblies (DAGCon) Chin et al., Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data, Nature Methods (2013) In de novo genome assembly, the longest reads are used as “backbone/seed” reads
Same “isoform hit” criteria – But does not require each read to be fully aligned – Each non-FL read can belong to multiple clusters Chin et al., Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data, Nature Methods (2013)
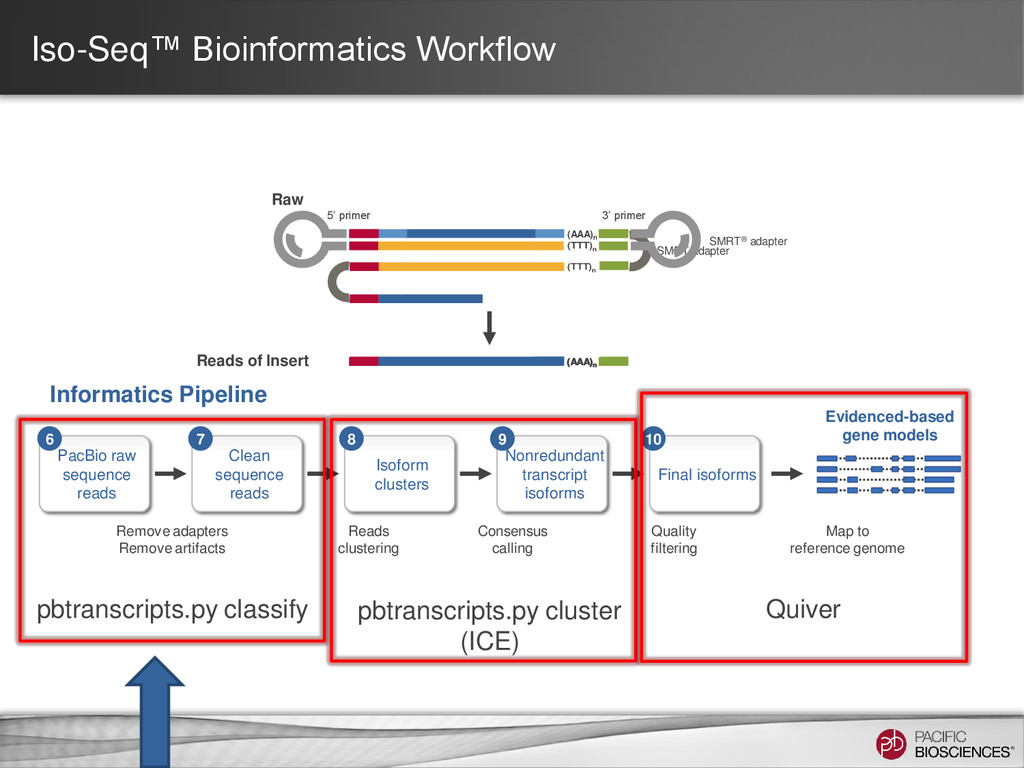
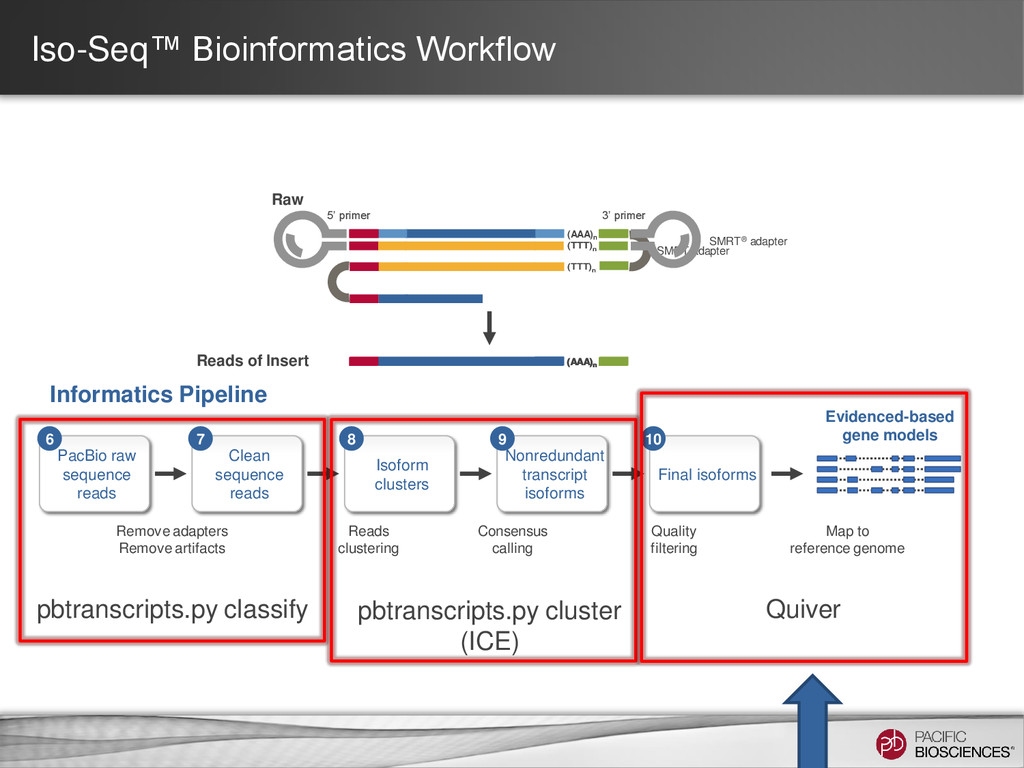
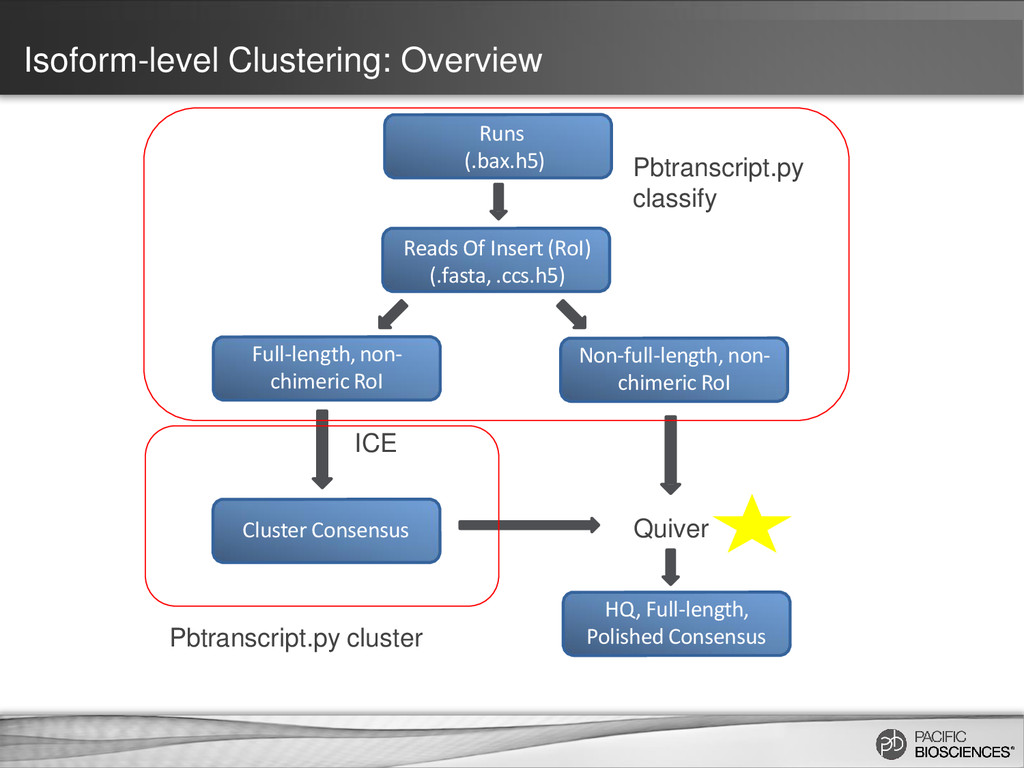
for isoform identification to improve accuracy – Better read classification (full-length vs. non-full-length, chimeric vs. non-chimeric) – Incorporation of non-full-length reads to predict consensus isoforms and for Quiver polishing • Usability enhancements within SMRT Portal UI supporting simplified analysis setup – Support for custom primers for targeted Iso-Seq projects – UI parameters tuning options for Quiver – Human-readable annotations for predicted consensus isoforms – Support for cDNA samples with no polyA tails • Performance enhancements – Faster job execution for analysis using ICE and polish (up to 3x) – Less memory usage (up to 8x) (*Note: Performance not guaranteed for custom primers) 35 https://github.com/PacificBiosciences/cDNA_primer/wiki
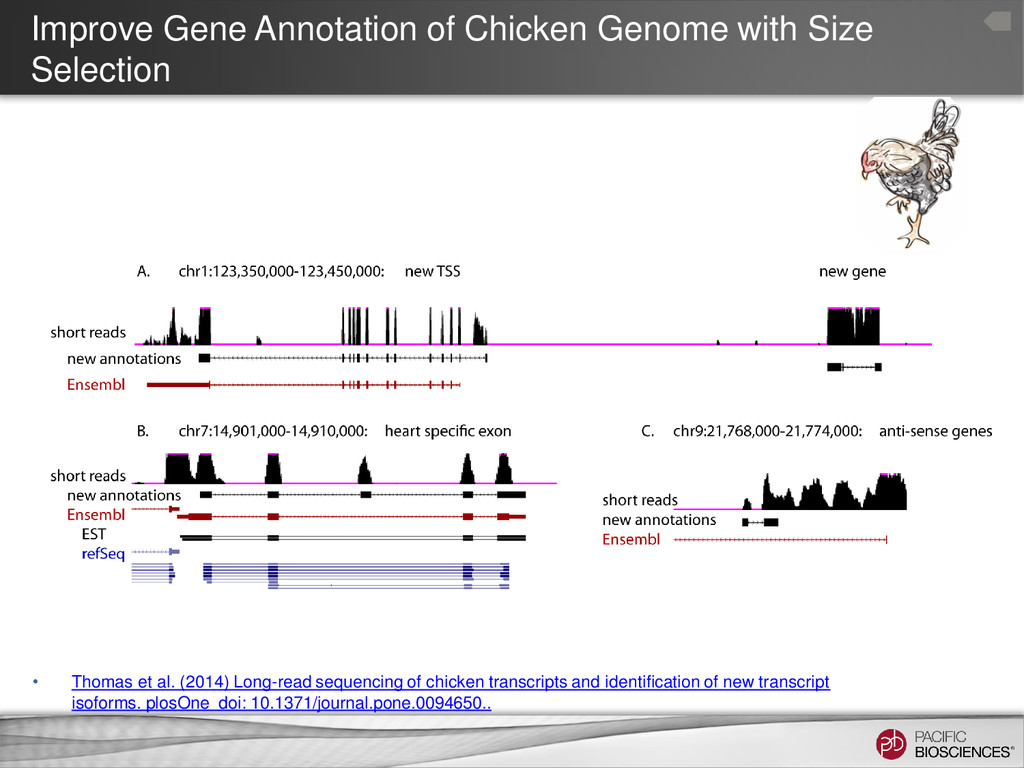
Thomas et al. (2014) Long-read sequencing of chicken transcripts and identification of new transcript isoforms. plosOne doi: 10.1371/journal.pone.0094650..
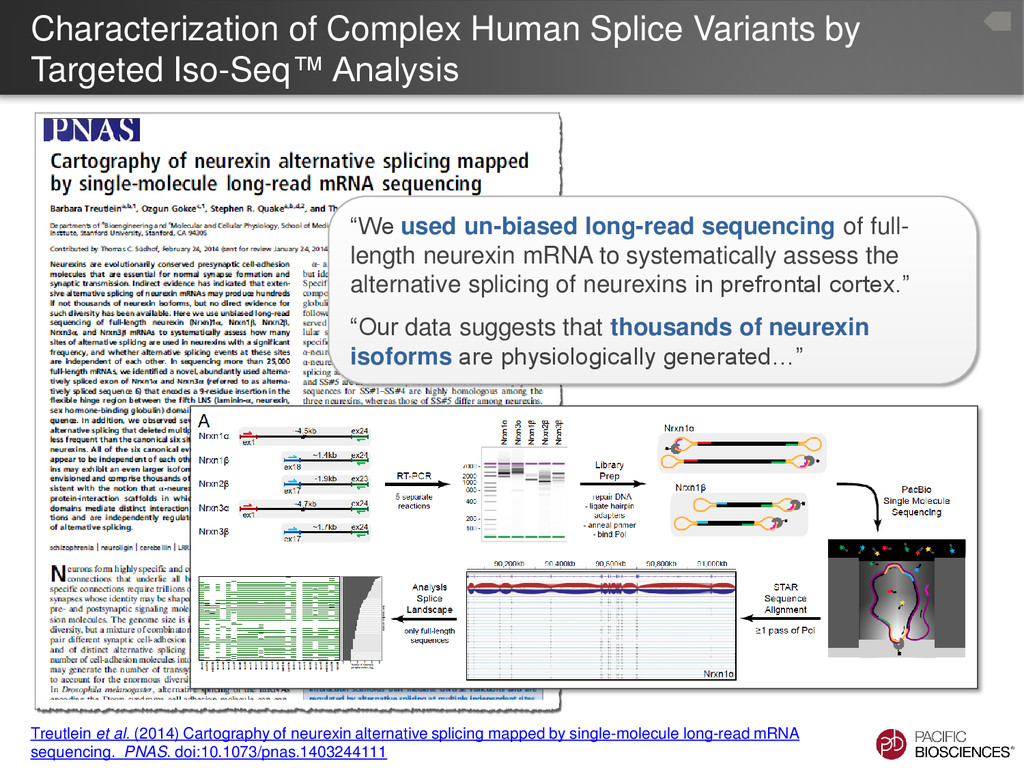
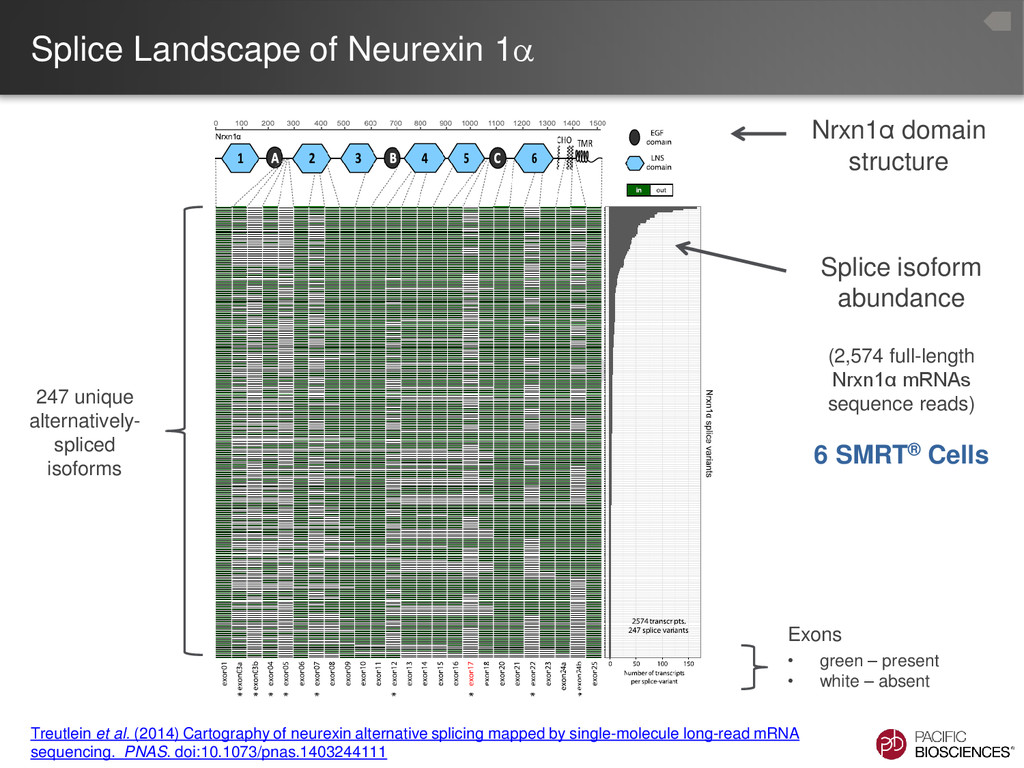
Treutlein et al. (2014) Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. PNAS. doi:10.1073/pnas.1403244111 “We used un-biased long-read sequencing of full- length neurexin mRNA to systematically assess the alternative splicing of neurexins in prefrontal cortex.” “Our data suggests that thousands of neurexin isoforms are physiologically generated…”
repertoires correlates with the cellular complexity of neuronal tissues, and a specific subset of isoforms is enriched in a purified cell type.” “Our analysis defines the molecular diversity of a critical synaptic receptor and provides evidence that neurexin diversity is linked to cellular diversity in the nervous system.” Numbers indicate total number of isoforms identified by PacBio® sequencing
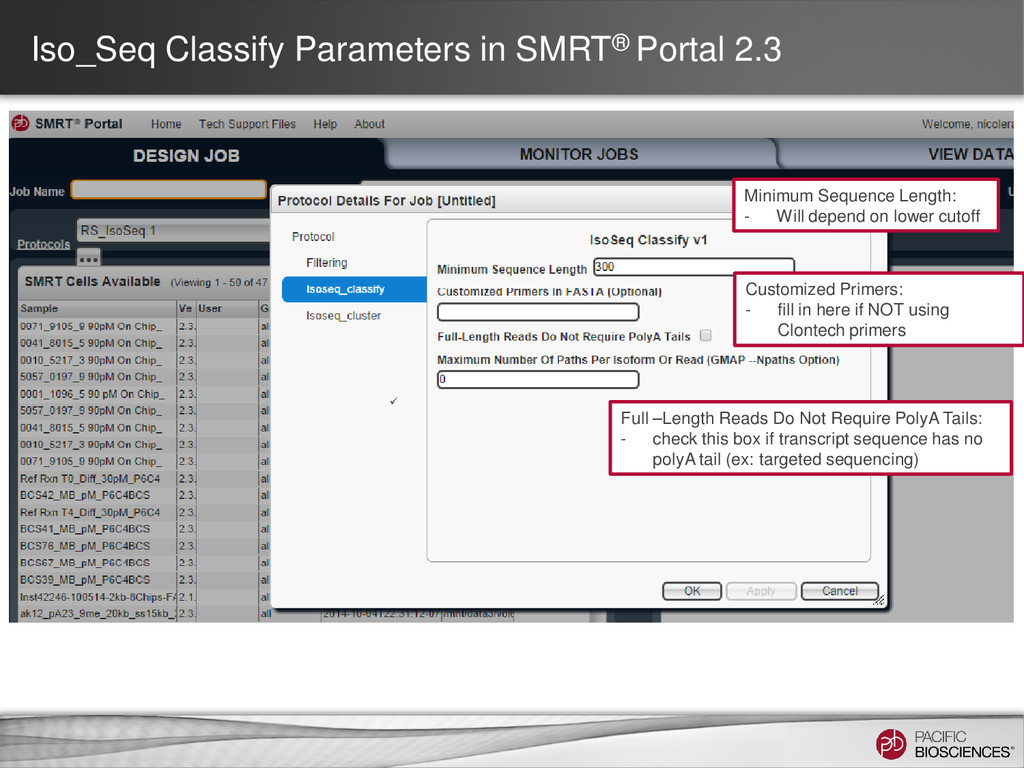
- Will depend on lower cutoff Full –Length Reads Do Not Require PolyA Tails: - check this box if transcript sequence has no polyA tail (ex: targeted sequencing) Customized Primers: - fill in here if NOT using Clontech primers
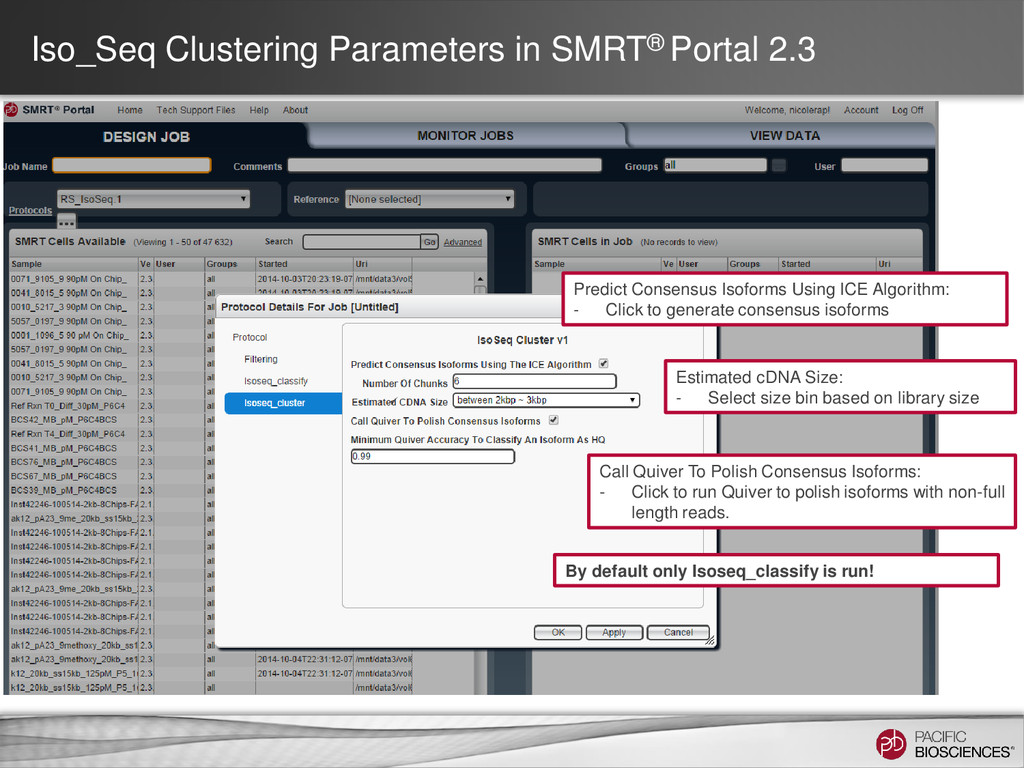
Using ICE Algorithm: - Click to generate consensus isoforms Call Quiver To Polish Consensus Isoforms: - Click to run Quiver to polish isoforms with non-full length reads. Estimated cDNA Size: - Select size bin based on library size By default only Isoseq_classify is run!
noncoding RNA acts competitively to regulate stem cell biology Thursday, October 16, 3:45 p.m. – 4:10 p.m. Vittorio Sebastiano, Ph.D., Stem Cell Biology and Regenerative Medicine Institute, Stanford School of Medicine
providers of long read sequencing solutions … “ Michael Snyder, Ph.D. Professor and Chair of Genetics Director, Stanford Center for Genomics and Personalized Medicine Stanford University
the human genome – novel insights with long read PacBio sequencing Tuesday, October 21, 12:30 p.m. – 2:00 p.m. Iso-Seq presentation by: Hagen Tilgner, Snyder Lab, Stanford Poster presentations: Monday (2:00 – 3:00 p.m.) 1627M: Full-length, single molecule whole transcriptome sequencing reveals alternative 5’- start sites, spliceoforms, and poly(A) addition signal sequences. David Munroe, NCI Tuesday (3:00 – 4:00 p.m.) 552T: Complex alternative splicing patterns in human hematopoietic cell subpopulations revealed by third-generation long reads Anne Deslattes Mays, Georgetown Univ, Lombardi Cancer Center
Pacific Biosciences, the Pacific Biosciences logo, PacBio, SMRT, SMRTbell, and Iso-Seq are trademarks of Pacific Biosciences in the United States and/or other countries. All other trademarks are the sole property of their respective owners.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}