of California, Inc. All rights reserved. For Research Use Only. Not for use in diagnostic procedures. Richard J Hall / 10/9/2014 Beyond Bacteria, Assembling >100 Mb Genomes Using Only PacBio® Sequence and Hybrid Assembly Methods
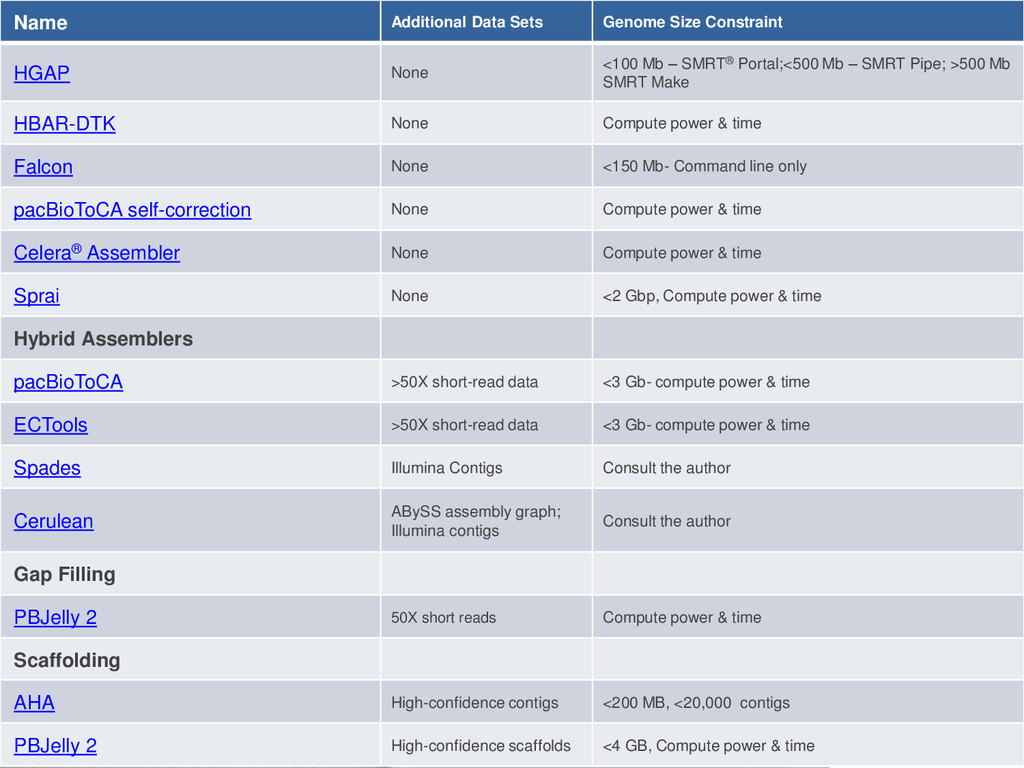
Mb – SMRT® Portal;<500 Mb – SMRT Pipe; >500 Mb SMRT Make HBAR-DTK None Compute power & time Falcon None <150 Mb- Command line only pacBioToCA self-correction None Compute power & time Celera® Assembler None Compute power & time Sprai None <2 Gbp, Compute power & time Hybrid Assemblers pacBioToCA >50X short-read data <3 Gb- compute power & time ECTools >50X short-read data <3 Gb- compute power & time Spades Illumina Contigs Consult the author Cerulean ABySS assembly graph; Illumina contigs Consult the author Gap Filling PBJelly 2 50X short reads Compute power & time Scaffolding AHA High-confidence contigs <200 MB, <20,000 contigs PBJelly 2 High-confidence scaffolds <4 GB, Compute power & time
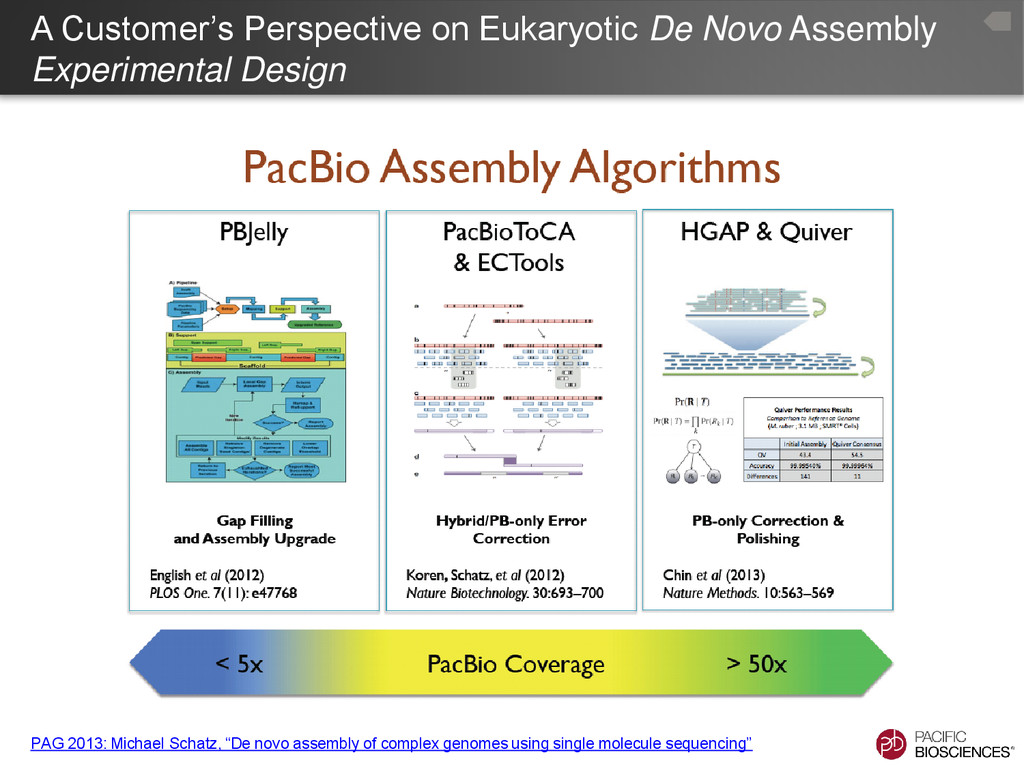
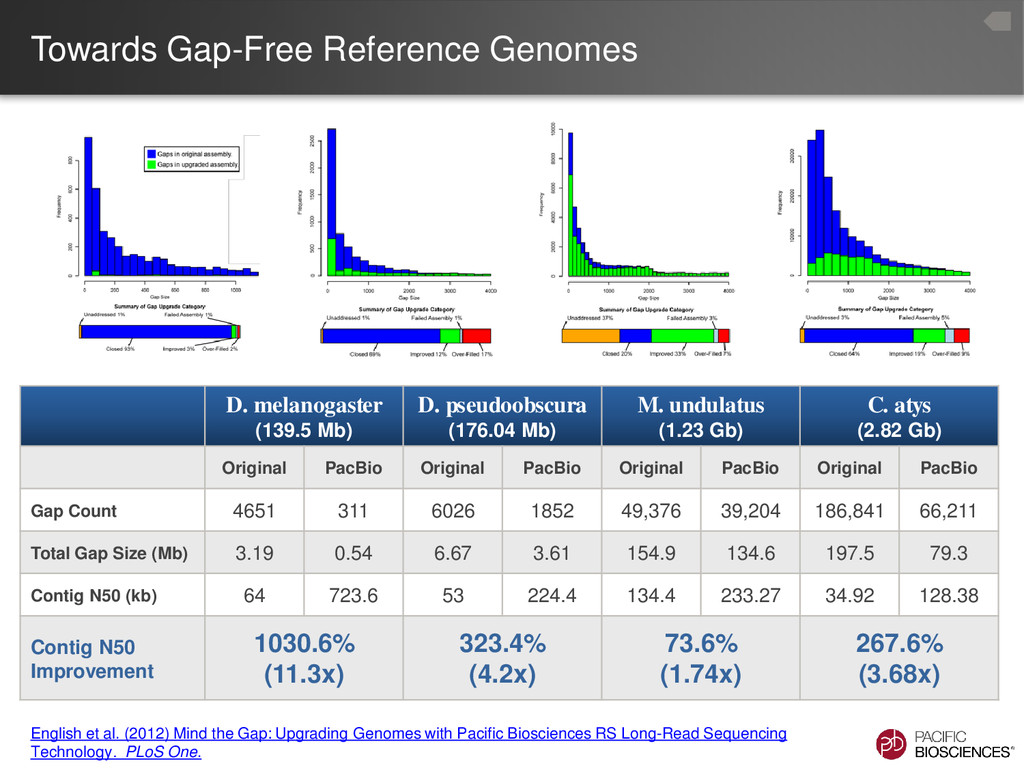
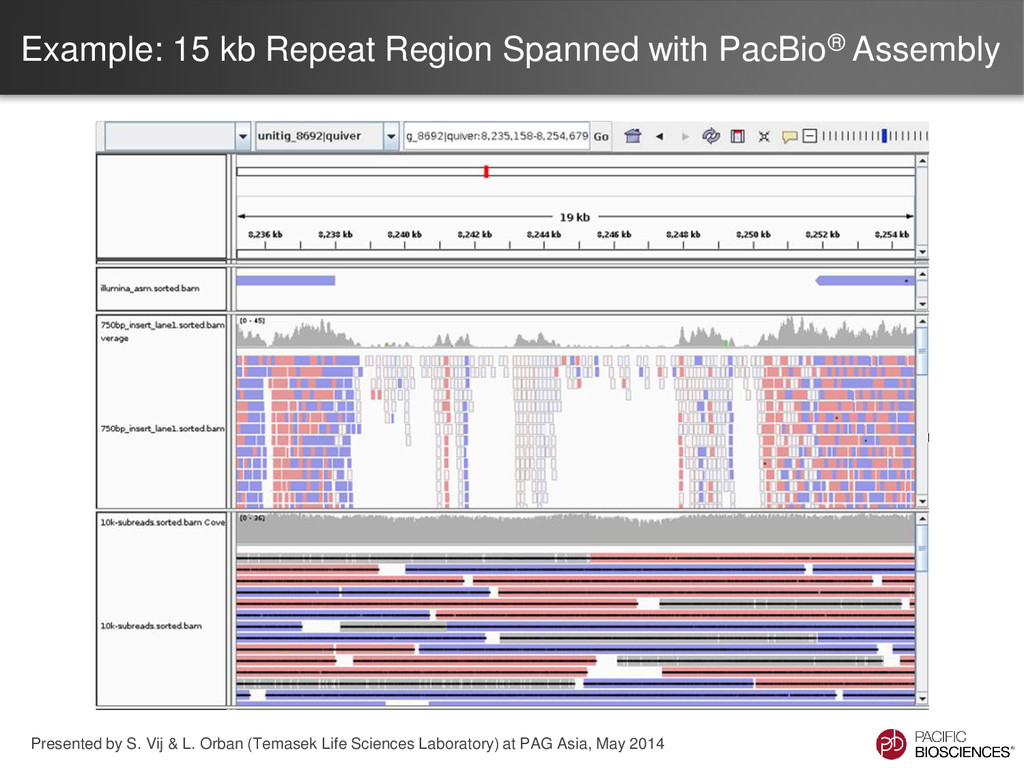
the genome upgrading tool. – PBHoney - the structural variation discovery tool • Advantages – Make use of very low-coverage PacBio® data – Significant improvements for well-studied assemblies – Reasonable computation times, even for large genomes • Disadvantages – Requires a ‘good’ draft genome – Limited by the current assembly – Will not correct for missing data in the current assembly, due to the limitations of the technologies used 7
to medium coverage PacBio® data – More computationally efficient that correcting with short reads (PBcR - http://wgs-assembler.sourceforge.net/wiki/index.php?title=PBcR) – Limited largely by computational resources, large genomes • Disadvantages – Does not overcome all the limitations of short-read assembly 10
Pacific Biosciences, the Pacific Biosciences logo, PacBio, SMRT, and SMRTbell are trademarks of Pacific Biosciences in the United States and/or other countries. All other trademarks are the sole property of their respective owners.
of California, Inc. All rights reserved. For Research Use Only. Not for use in diagnostic procedures. James Drake – Bioinformatics Workshop 10/2014 SMRT® Make
single file, but desire to modularize is overwhelming. – Chunking, i.e., splitting the data up for parallel processing. – Defining cluster submission pragmas (e.g., qsub calls) – Data filtering – Reports • Provide the general purpose use case, user customizes to fit their needs. • Minimal parameters • Serves as a record of the experiment • Easily reproducible by others, just share your make file. • Create self-contained make files by simply `cat`ing them together.
to submit jobs to QUEUE ?= huasm # Size of the genome GENOME_SIZE ?= 700000000 # Splits data into this many chunks, each chunk processed independently CHUNK_SIZE ?= 15 # How many threads a process will use (also how many SGE slots will be requested) NPROC ?= 32 # Local temp root directory, must have write access and a decent amount of space (~100GB) LOCALTMP ?= /scratch Parameters Initialization Recipes Typically run in bottom-up order
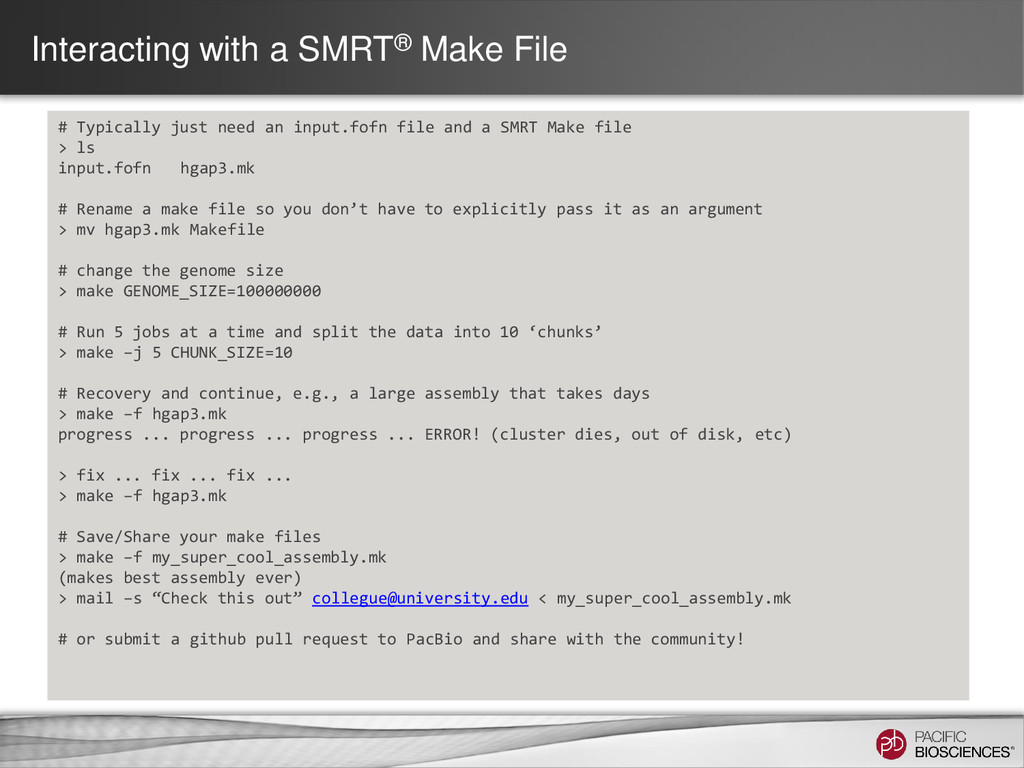
an input.fofn file and a SMRT Make file > ls input.fofn hgap3.mk # Rename a make file so you don’t have to explicitly pass it as an argument > mv hgap3.mk Makefile # change the genome size > make GENOME_SIZE=100000000 # Run 5 jobs at a time and split the data into 10 ‘chunks’ > make –j 5 CHUNK_SIZE=10 # Recovery and continue, e.g., a large assembly that takes days > make –f hgap3.mk progress ... progress ... progress ... ERROR! (cluster dies, out of disk, etc) > fix ... fix ... fix ... > make –f hgap3.mk # Save/Share your make files > make –f my_super_cool_assembly.mk (makes best assembly ever) > mail –s “Check this out” [email protected] < my_super_cool_assembly.mk # or submit a github pull request to PacBio and share with the community!
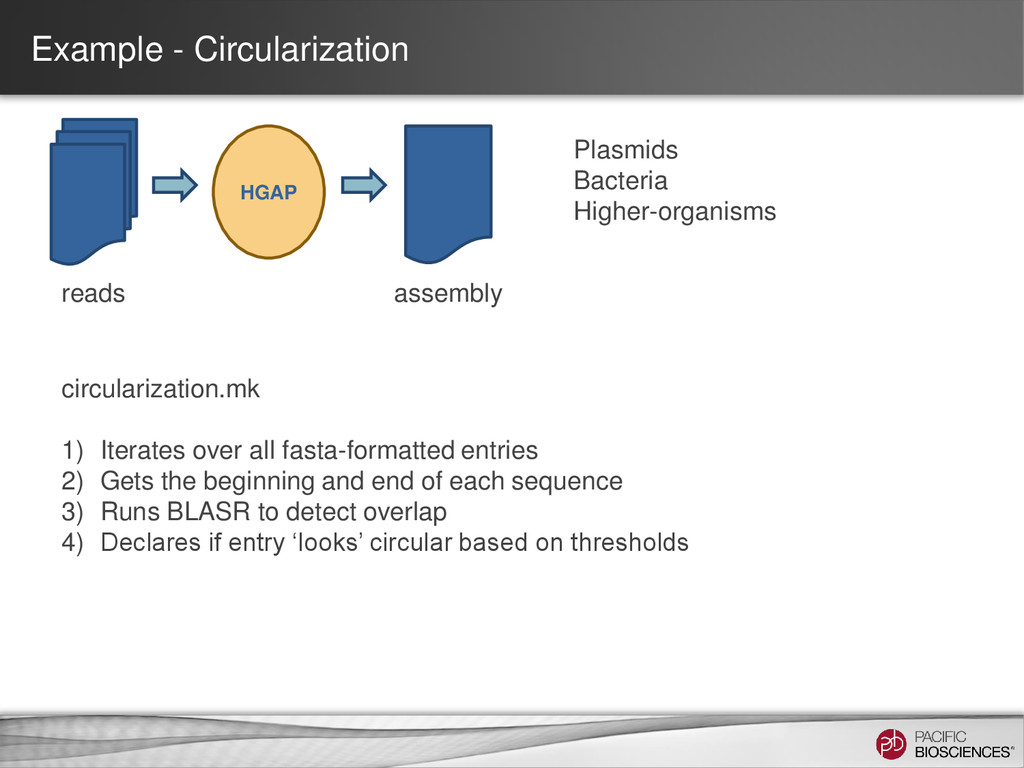
1) Iterates over all fasta-formatted entries 2) Gets the beginning and end of each sequence 3) Runs BLASR to detect overlap 4) Declares if entry ‘looks’ circular based on thresholds
Pacific Biosciences, the Pacific Biosciences logo, PacBio, SMRT, and SMRTbell are trademarks of Pacific Biosciences in the United States and/or other countries. All other trademarks are the sole property of their respective owners.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}