of California, Inc. All rights reserved. For Research Use Only. Not for use in diagnostic procedures. User Group Meeting – Menlo Park, October 2014 De Novo Assembly
Design and De Novo assembly using the PacBio® HGAP method After the training, you will be able to • Understand how the HGAP method works • Understand the coverage targets for de novo assembly with PacBio data • Import and parameterize an HGAP assembly job in SMRT® Portal • SMRT® Technology • PacBio® System Workflow
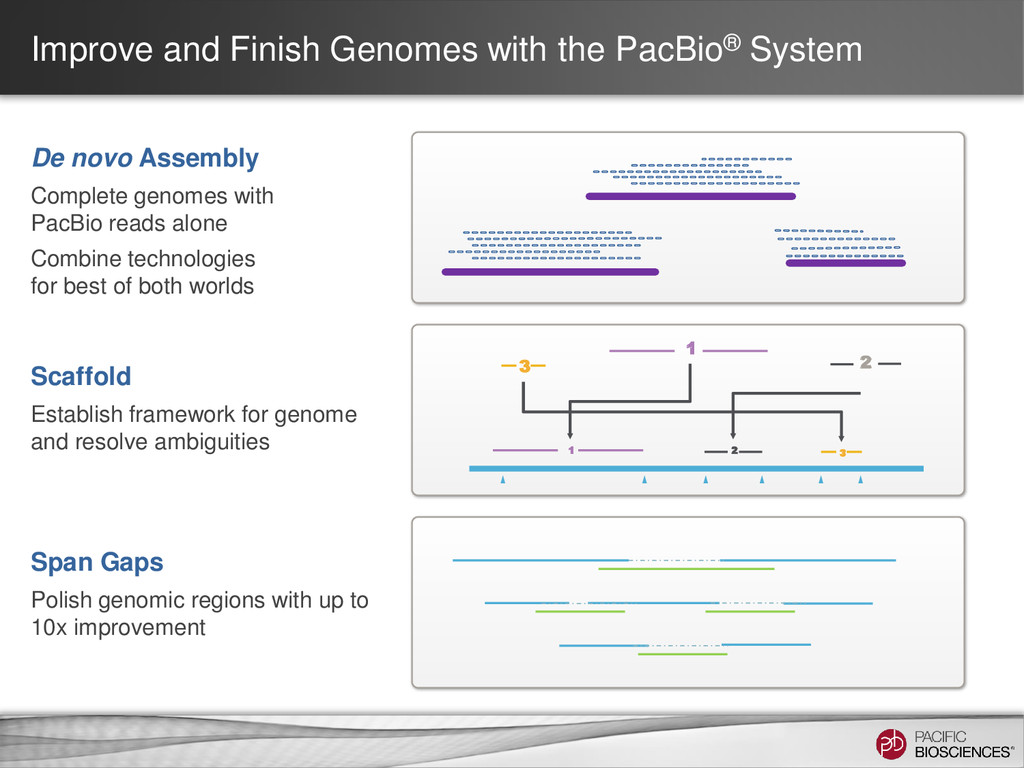
Assembly Complete genomes with PacBio reads alone Combine technologies for best of both worlds 2 3 2 3 1 1 Scaffold Establish framework for genome and resolve ambiguities Span Gaps Polish genomic regions with up to 10x improvement
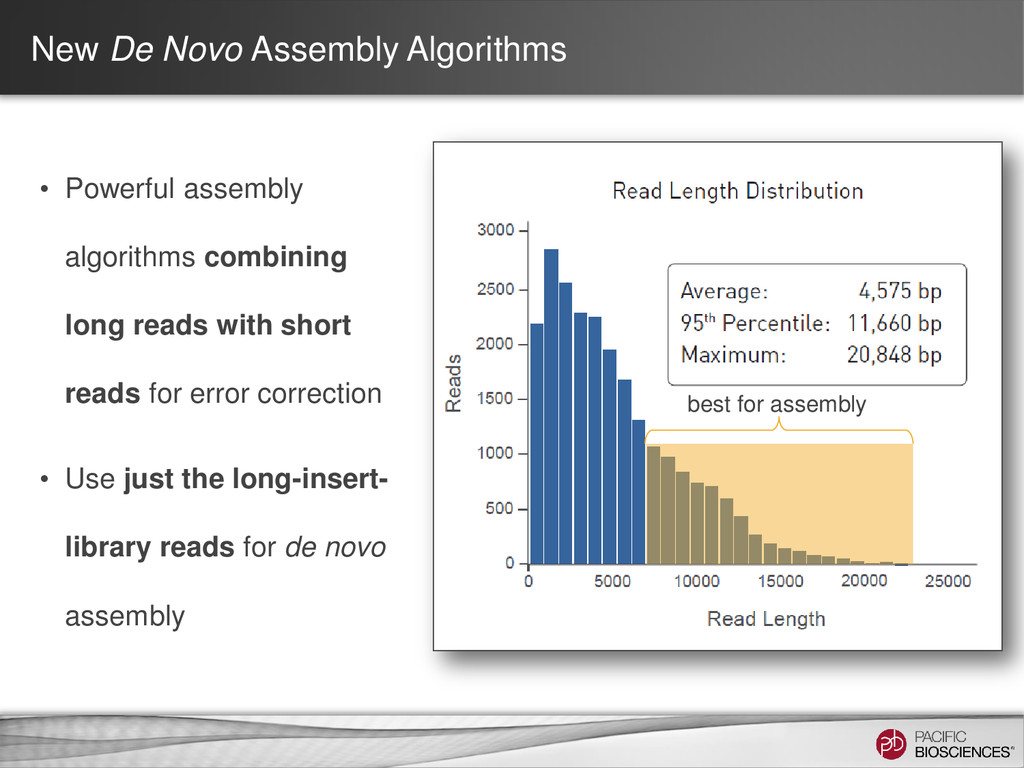
Sequencing reads with short reads for error correction • Requires multiple types of data, (at least) two library preps, different sequencing technologies…
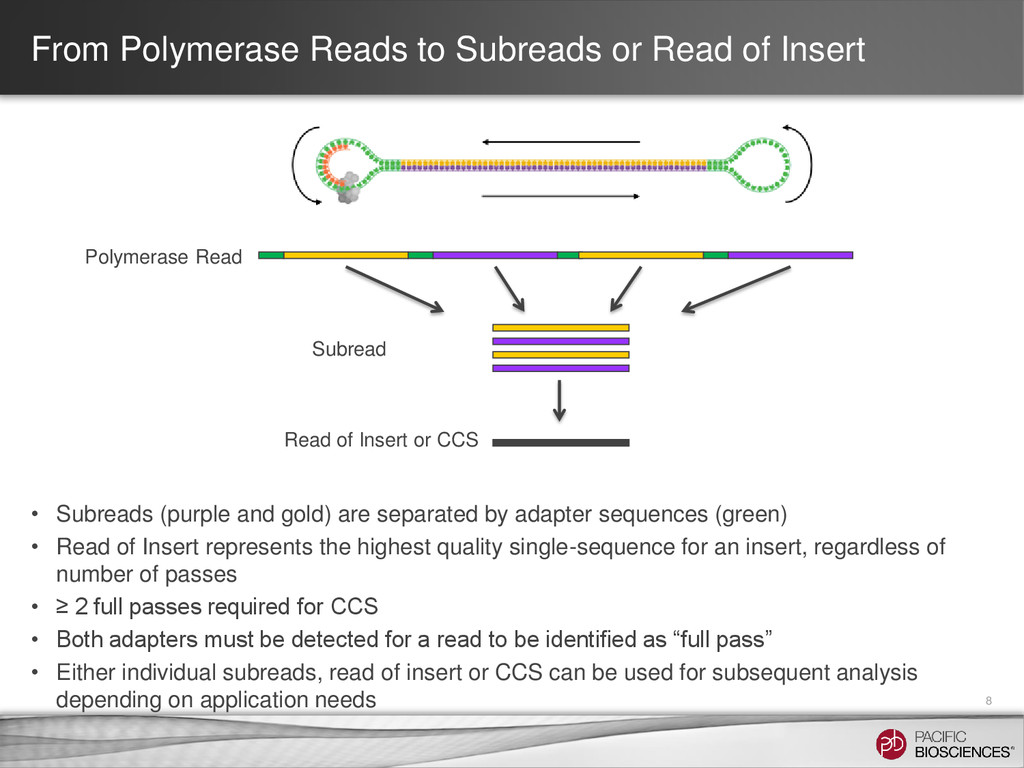
Subreads (purple and gold) are separated by adapter sequences (green) • Read of Insert represents the highest quality single-sequence for an insert, regardless of number of passes • ≥ 2 full passes required for CCS • Both adapters must be detected for a read to be identified as “full pass” • Either individual subreads, read of insert or CCS can be used for subsequent analysis depending on application needs Polymerase Read Subread Read of Insert or CCS 8
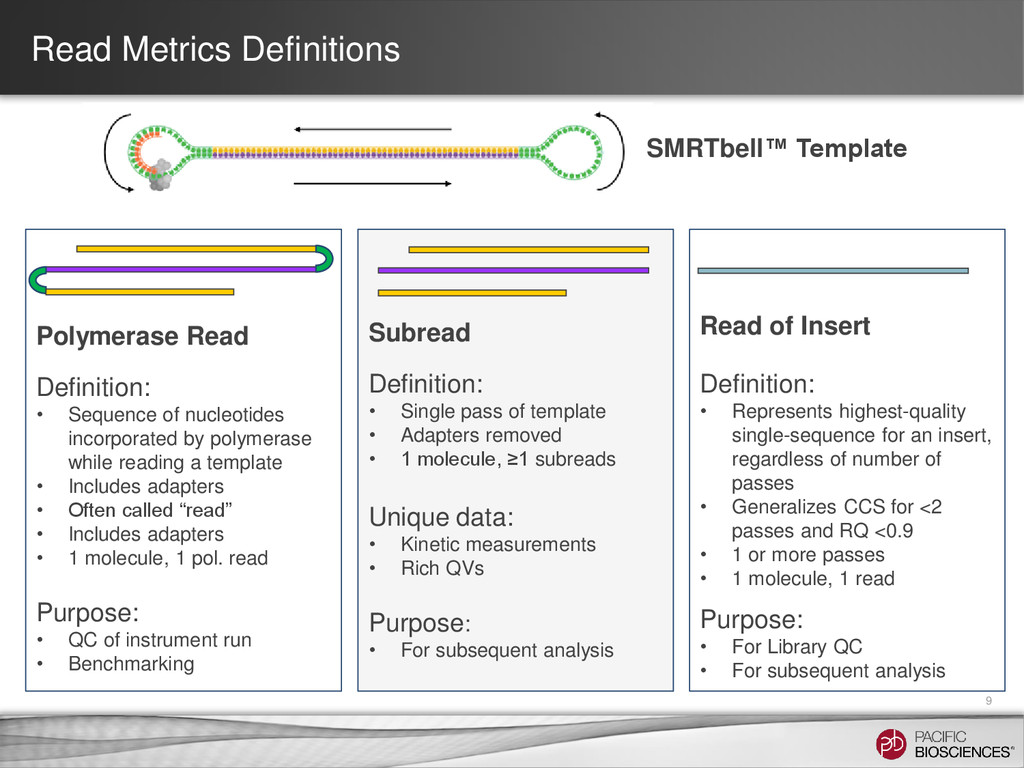
insert, regardless of number of passes • Generalizes CCS for <2 passes and RQ <0.9 • 1 or more passes • 1 molecule, 1 read Purpose: • For Library QC • For subsequent analysis Subread Definition: • Single pass of template • Adapters removed • 1 molecule, ≥1 subreads Unique data: • Kinetic measurements • Rich QVs Purpose: • For subsequent analysis Polymerase Read Definition: • Sequence of nucleotides incorporated by polymerase while reading a template • Includes adapters • Often called “read” • Includes adapters • 1 molecule, 1 pol. read Purpose: • QC of instrument run • Benchmarking Read Metrics Definitions SMRTbell™ Template 9
Read Length Mapped Subread Length 4 kb 900 bp Mapped Polymerase Read Length Measure of ZMW sequencing productivity and read length Upper bound by speed and fidelity of the polymerase and movie time Mapped Subread Length Measure of scientifically applicable sequence Upper bound by insert size and loading effects
of contigs – N50: Equal to the size of the contig found if you sort contigs by size and walk to the contig that represents 50% of the total sequence − N50 = 10 bp − Mean contig length = 3 bp – Max contig size • Limitation of these metrics: – They do not capture information about assembly accuracy! − Large scale misassemblies − Base-level errors – There might be more than one chromosome (plasmid, phage, and so on) – Contaminants may contribute to a contig (such as a cloning vector) 11 10 4 1 1 1 1
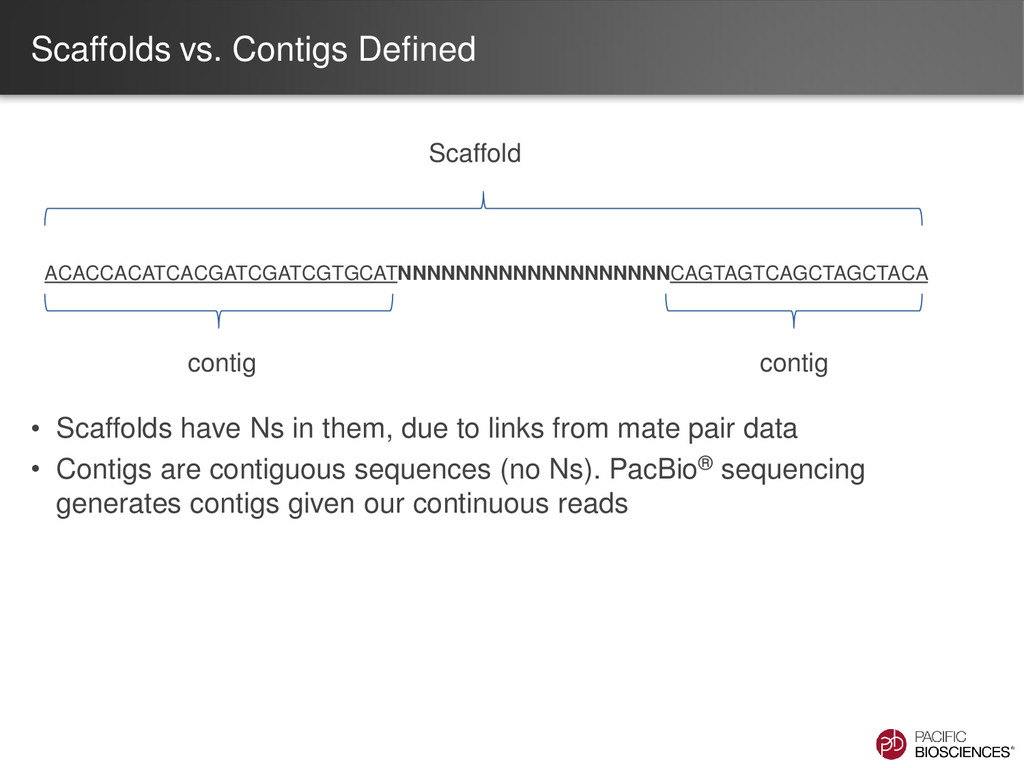
due to links from mate pair data • Contigs are contiguous sequences (no Ns). PacBio® sequencing generates contigs given our continuous reads ACACCACATCACGATCGATCGTGCATNNNNNNNNNNNNNNNNNNNCAGTAGTCAGCTAGCTACA contig contig Scaffold
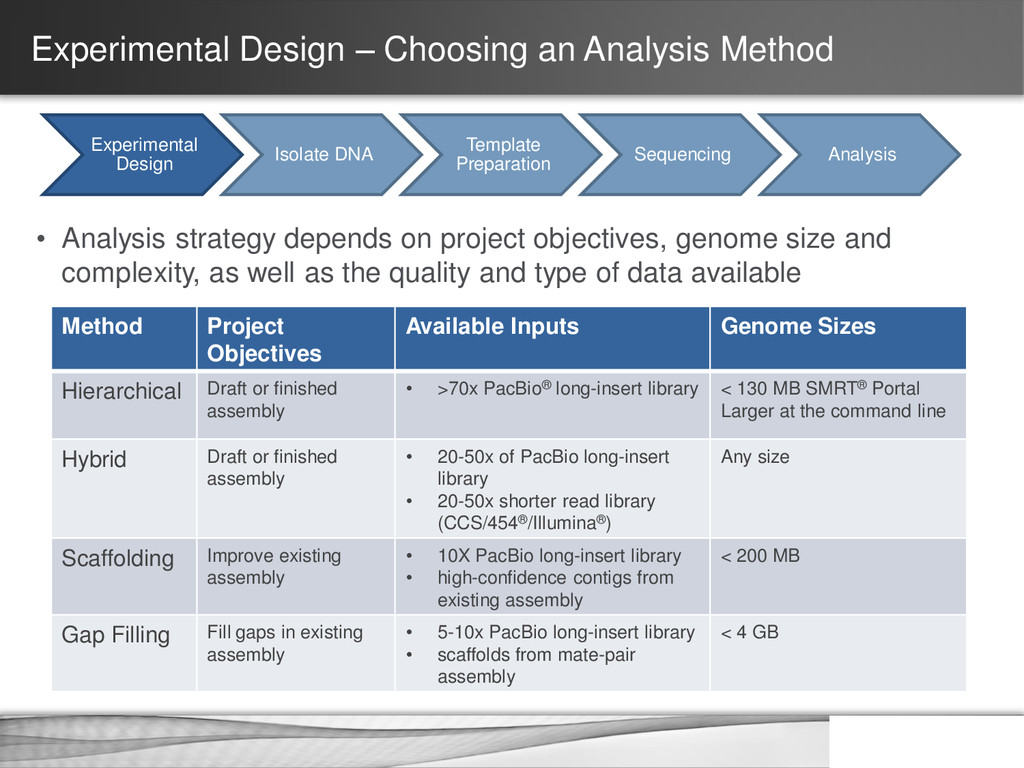
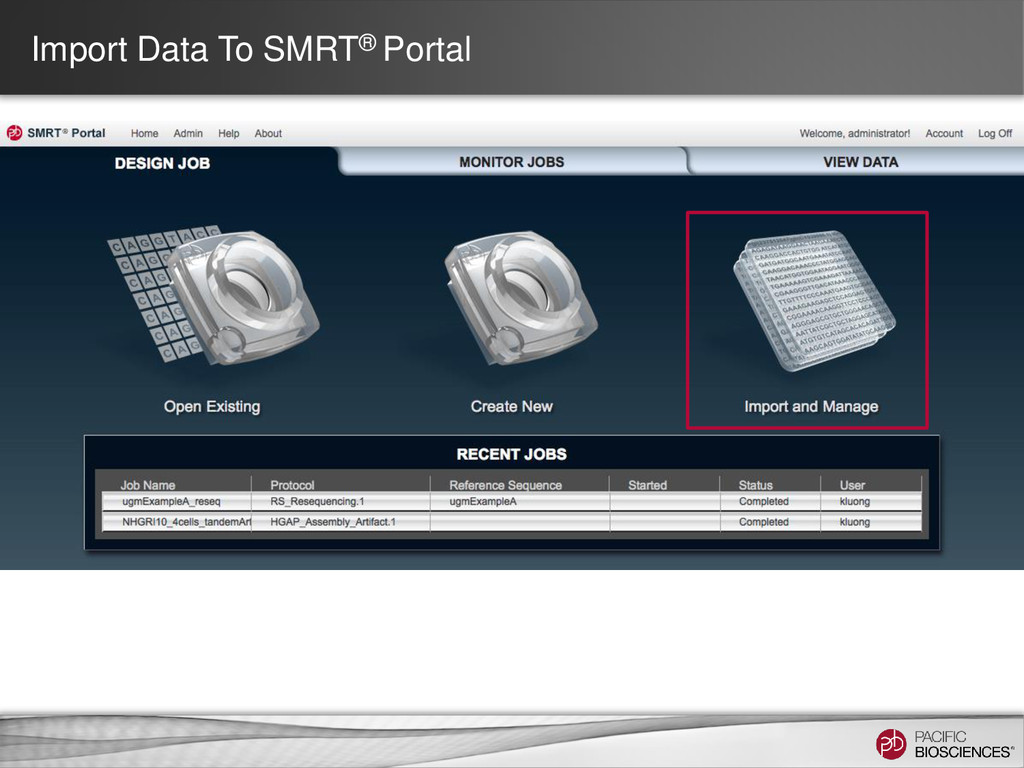
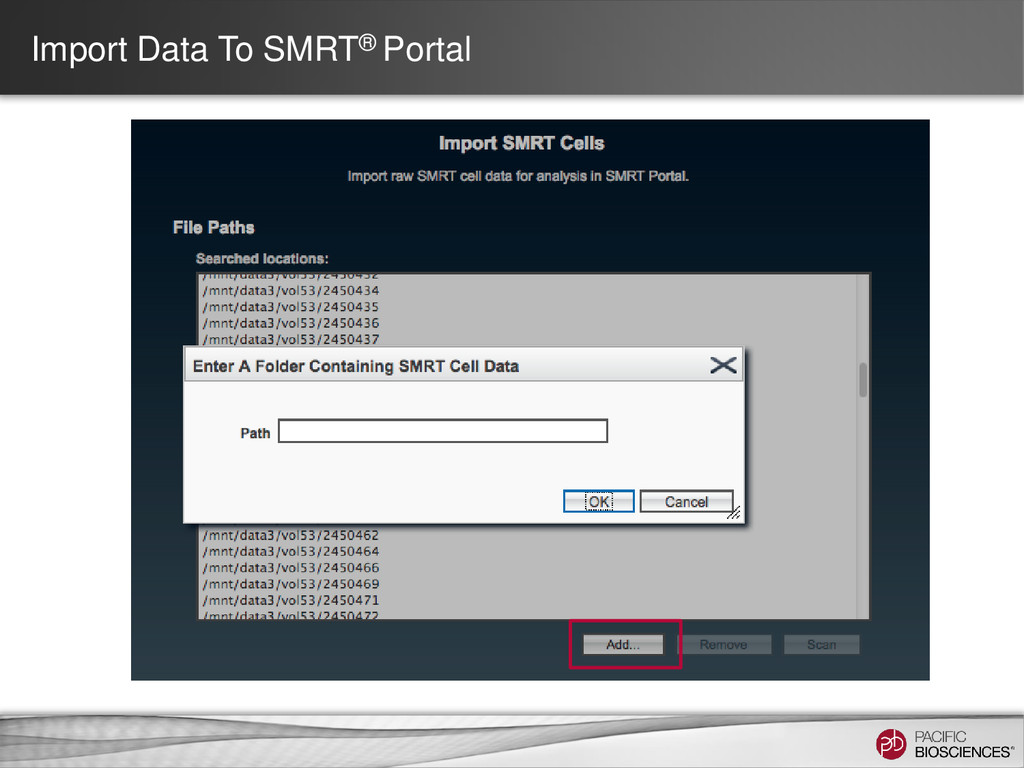
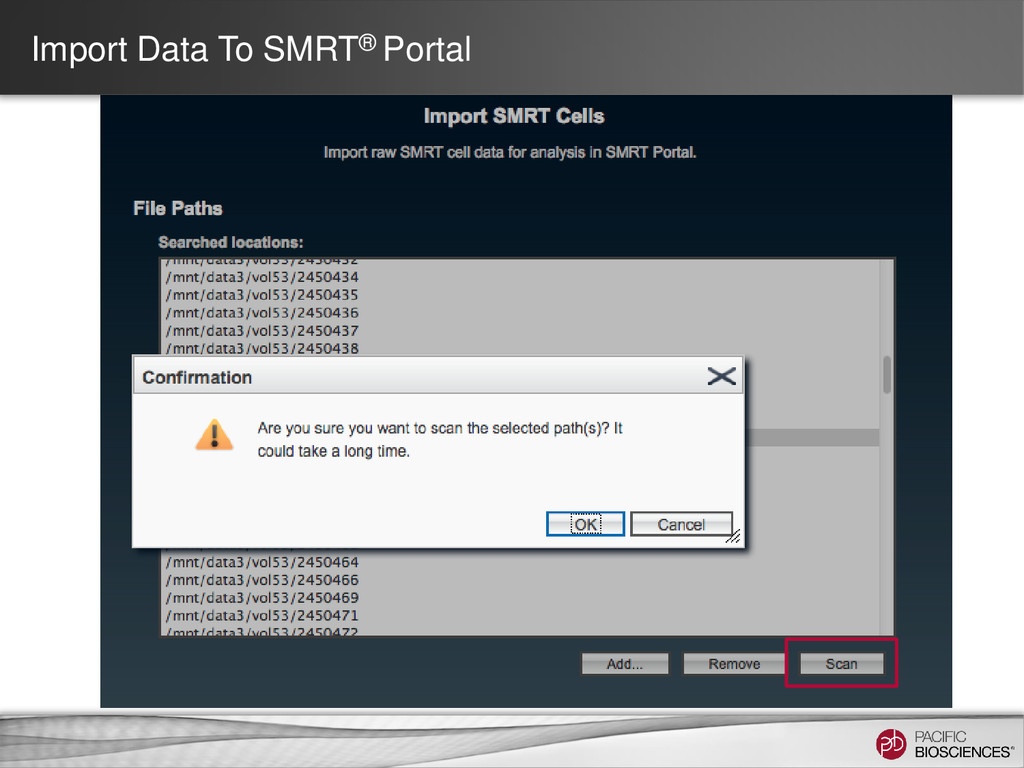
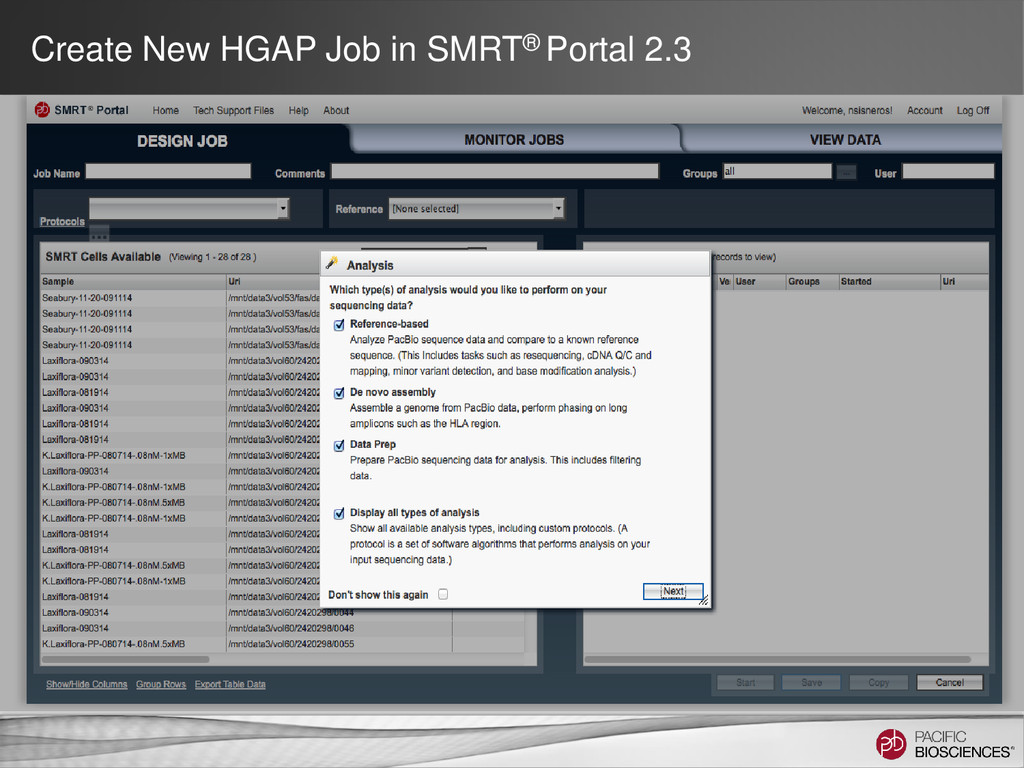
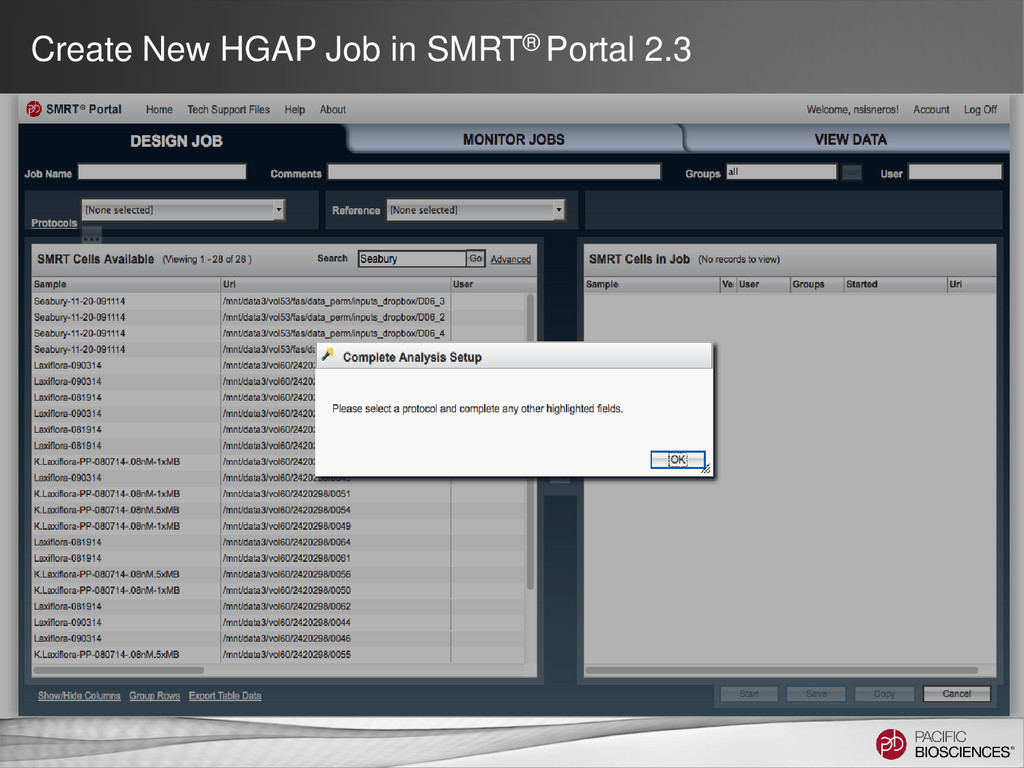
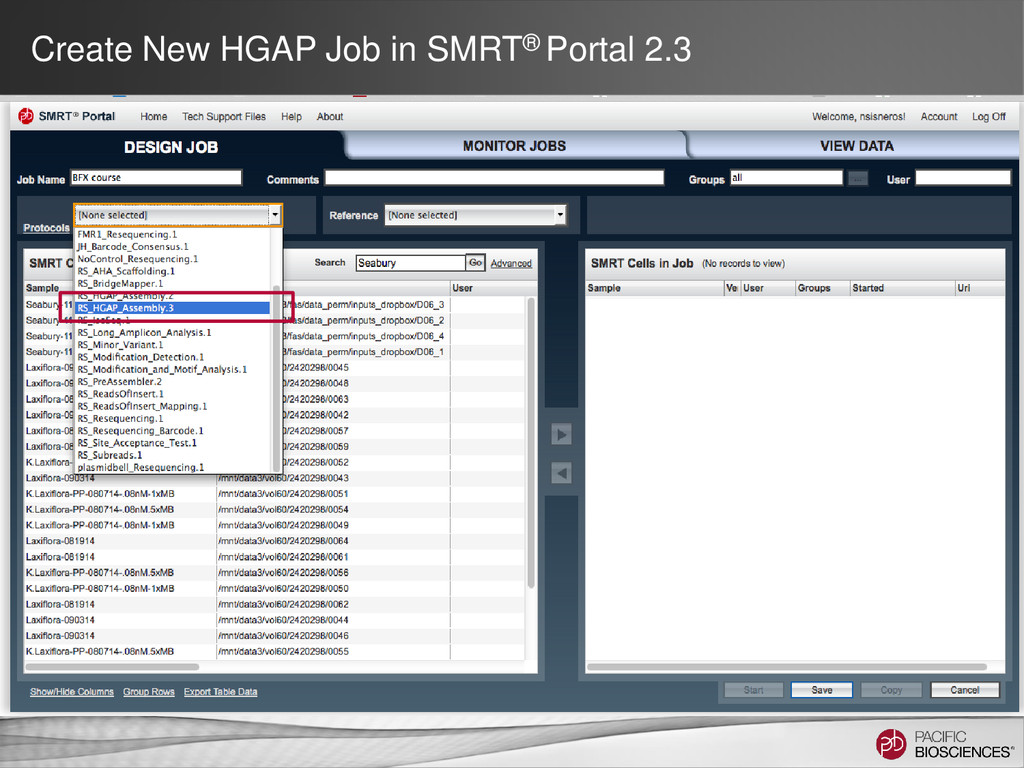
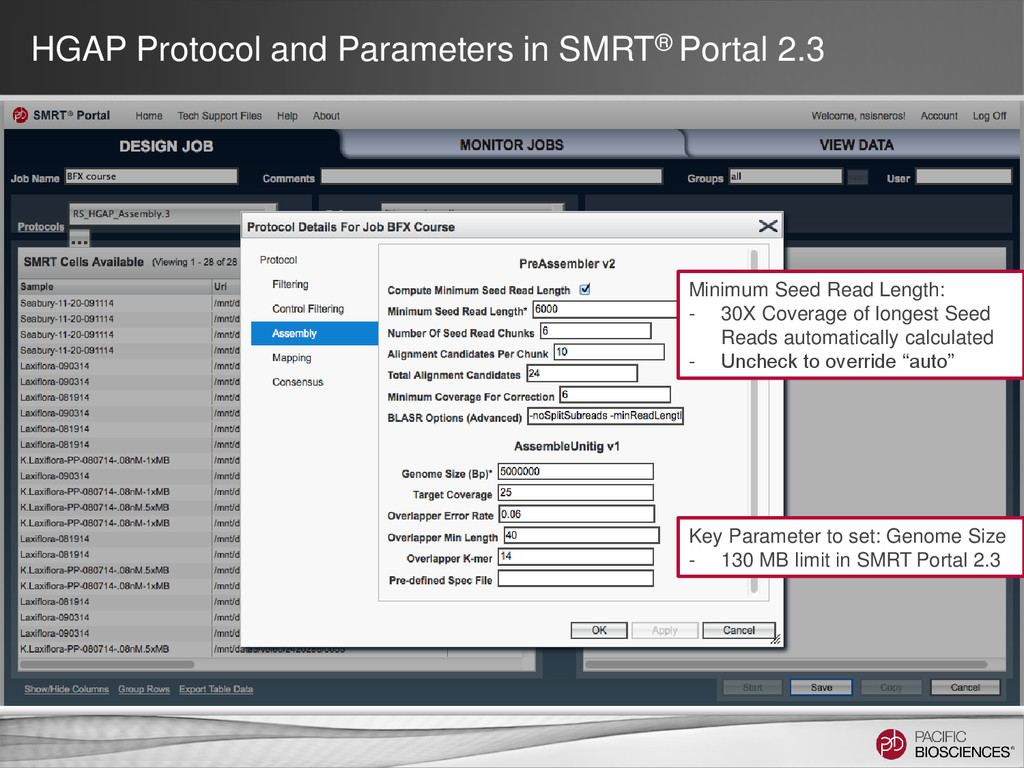
DNA Template Preparation Sequencing Analysis • Analysis strategy depends on project objectives, genome size and complexity, as well as the quality and type of data available Method Project Objectives Available Inputs Genome Sizes Hierarchical Draft or finished assembly • >70x PacBio® long-insert library < 130 MB SMRT® Portal Larger at the command line Hybrid Draft or finished assembly • 20-50x of PacBio long-insert library • 20-50x shorter read library (CCS/454®/Illumina®) Any size Scaffolding Improve existing assembly • 10X PacBio long-insert library • high-confidence contigs from existing assembly < 200 MB Gap Filling Fill gaps in existing assembly • 5-10x PacBio long-insert library • scaffolds from mate-pair assembly < 4 GB
Preparation Sequencing Analysis • Repetitive Content - One of the biggest challenges with De Novo Assembly. - Solution: work with insert sizes that can span repeats and indentify unique anchoring sequencing on each side of the repeat.
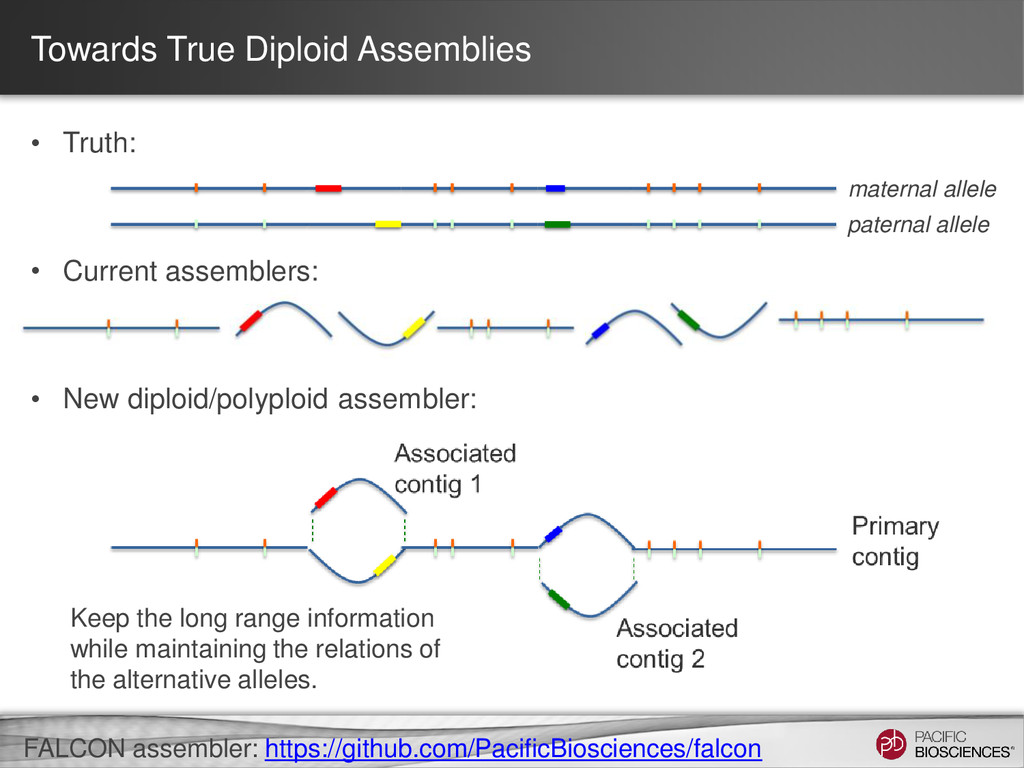
Haploid Genomes. – Diploid with little structural variation between the chromosomes then a haploid approach can work. – Structural heterozygosity appears as separate contigs. • Select Strains to minimize heterozygosity – This helps facilitate assembly. – Use inbred lines – Double Haploid strains • Diploid or multiploid genomes – Using a haploid assembler leads to fragmented assemblies. – Consider Falcon (experimental Code) or Celera Assembler can be configured to favor merging haplotypes. 26
New diploid/polyploid assembler: maternal allele paternal allele Keep the long range information while maintaining the relations of the alternative alleles. FALCON assembler: https://github.com/PacificBiosciences/falcon
assemblies – Sensitive to the quality of the starting assembly. – Missassemblies in the scaffolds can result in improper alignments and incorrectly-filled or unfillable gaps. 28 Scaffolds with gaps PacBio long reads Filled in scaffold Gap Filling: Using PacBio® CLR to fill gaps in existing mate-pair-based scaffolds
generation of the longest reads possible. • Sample quality is critical to maximize potential performance • No amplification step during library preparation • Recommendations: – Take care during extraction to avoid gDNA damage & avoid contaminants – Use extraction methods or kits that produce very high molecular weight gDNA – If contaminants are present, purify starting DNA material prior to library prep – Accurately quantify and qualitatively evaluate gDNA – Include DNA-damage-repair step in library prep 29 Experimental Design Isolate DNA Template Preparation Sequencing Analysis
Nederbragt Blog) 32 http://flxlexblog.wordpress.com/2013/06/19/longing-for-the-longest-reads-pacbio-and-bluepippin/ “The plot shows that the BluePippin prep indeed had the desired effect: the reads are much longer.” N50 4,041bp 8,820bp
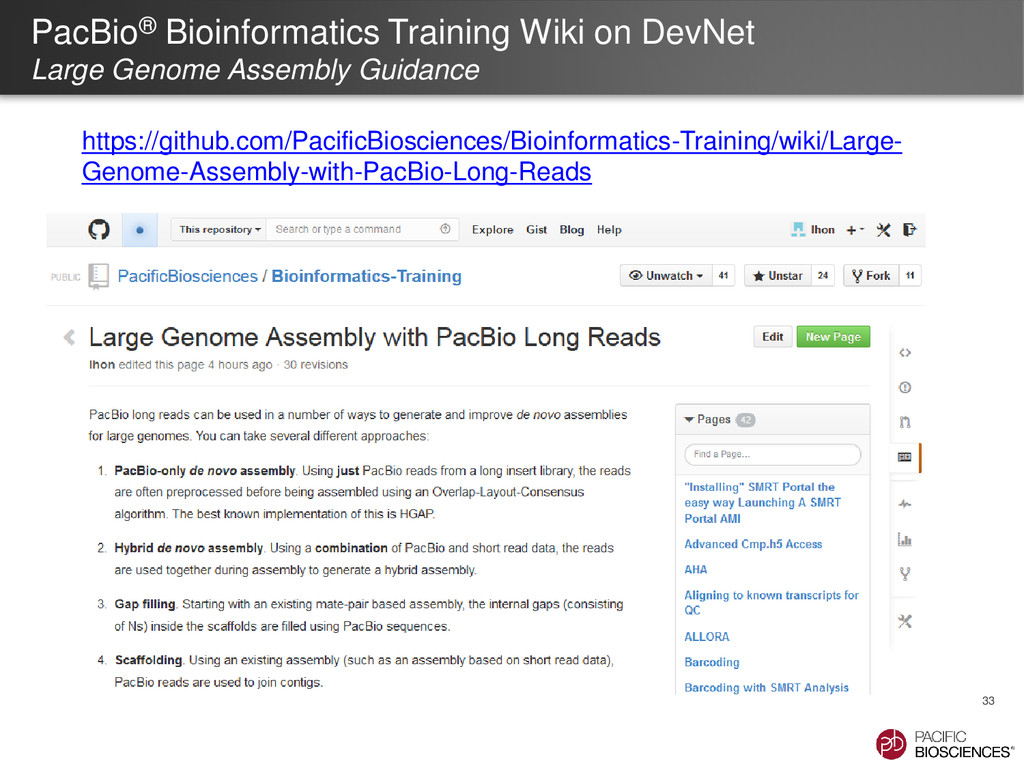
of presentations, posters and other de novo assembly resources available through PacBio’s website (www.pacb.com/denovo) • Protocols, Technical & Application Notes available through Customer Portal • DevNet – HGAP Reference Implementation: http://www.smrtcommunity.com/Share/Code?id=a1q70000000H2qRAAS – Quiver: www.pacbiodevnet.com/quiver – Bacterial Assembly and Epigenetic Analysis Training Web Video http://www.pacificbiosciences.com/Tutorials/Bacterial_Assembly_Epigenetic_Analysis_HGA P/story_html5.html • Additional information on Assembly Tools – Celera® Assembler: http://sourceforge.net/apps/mediawiki/wgs- assembler/index.php?title=PacBioToCA – Allpaths-LG: http://www.broadinstitute.org/software/allpaths-lg/blog/ – PBJelly: http://sourceforge.net/p/pb-jelly/wiki/Home/ 34
Pacific Biosciences, the Pacific Biosciences logo, PacBio, SMRT, SMRTbell, and Iso-Seq are trademarks of Pacific Biosciences in the United States and/or other countries. All other trademarks are the sole property of their respective owners.
Pacific Biosciences, the Pacific Biosciences logo, PacBio, SMRT, SMRTbell, and Iso-Seq are trademarks of Pacific Biosciences in the United States and/or other countries. All other trademarks are the sole property of their respective owners.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}