of California, Inc. All rights reserved. For Research Use Only. Not for use in diagnostic procedures. Matthew Seetin, Roberto Lleras, and Richard Hall / June 16 & 18, 2015 Planning, Running, and Understanding the FALCON Genome Assembly Pipeline
software, virtual environments • Understand your cluster’s file system • Understand your cluster’s queuing system – Syntax, time and other resource limits • Understand your organism – Size, ploidy, heterozygosity, repeat content 3
Begins with all-by-all comparison of raw subreads, matches written to disk – Matches then read, sorted, and merged into error-corrected, preassembled reads – These preassembled reads are nearly as valuable as the final result! • FALCON is limited by file i/o capabilities – Lustre file system recommended – NFS can only handle 3-5 concurrent jobs during preassembly step – Highly repetitive genomes require quadratically more storage space – In exchange, 20x reduction in CPU time for human assembly
“SGE,” for running on a Sun Grid Engine cluster • Running locally only practical for toy genomes input_fofn = input.fofn • File of file names listing all input files input_type = raw #input_type = preads • Can restart job using preassembled reads to quickly test alternative downstream parameters 7
for seed reads used for initial mapping length_cutoff = 1000 • Use your longest 30x of coverage. Length correlates strongly with assembly quality # The length cutoff used for seed reads used for pre-assembled reads length_cutoff_pr = 1000 • Usually 0-5000 shorter than length_cutoff. Shorter number may increase contiguity at the risk of misassembly 8
= sge_option_pla = sge_option_fc = sge_option_cns = pa_concurrent_jobs = 1 ovlp_concurrent_jobs = 1 • These settings depend on your cluster setup; text is inserted after qsub command. • sge_option_da, sge_option_la, and pa_concurrent_jobs govern most expensive steps. 9
-e.70 -l500 -s500 ovlp_HPCdaligner_option = -v -dal4 -t32 -h60 -e.96 -l500 -s500 • Flags for Daligner • -dal4 specifies 4 serial calls to daligner command per job submitted to the queue • -l and -s set how many base pairs constitute the minimum local alignment and how frequently (in bases) these are recorded, respectively • -t suppresses k-mer frequency (suppress repeats) so as to limit memory usage • For more information: https://dazzlerblog.wordpress.com/ 10
= -x500 -s50 • Flags for how the read database is split up between jobs • -s specifies number of megabytes in each DB chunk: larger number generates a smaller number of longer jobs. • For more information: https://dazzlerblog.wordpress.com/ 11
--min_cov 4 --local_match_count_threshold 2 --max_n_read 200 --n_core 2 • Flags pertain to error correction of raw reads • These settings work fine for many assemblies, but max_n_read may need to be lowered in highly repetitive genomes. overlap_filtering_setting = --max_diff 100 --max_cov 100 -- min_cov 2 • -min_cov specifies minimum coverage in preassembled reads in order to continue to extend contig. Low values promote contiguity at expense of misassembly • -max_diff specifies the maximum difference in coverage between ends of a pread, which usually indicates a repeat 12
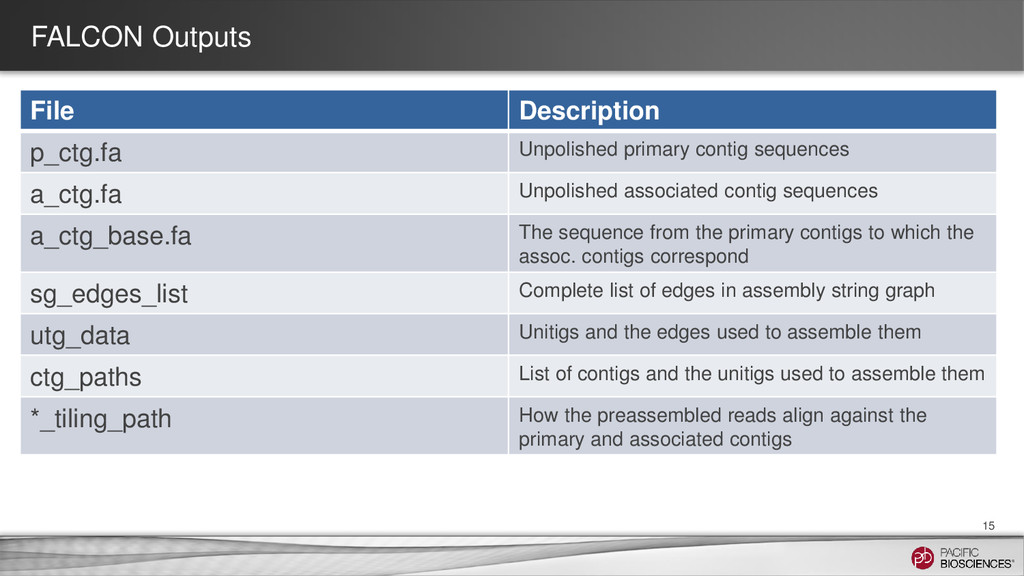
Unpolished associated contig sequences a_ctg_base.fa The sequence from the primary contigs to which the assoc. contigs correspond sg_edges_list Complete list of edges in assembly string graph utg_data Unitigs and the edges used to assemble them ctg_paths List of contigs and the unitigs used to assemble them *_tiling_path How the preassembled reads align against the primary and associated contigs 15
are unambiguously- resolved straight lines of edges • Consecutive edges usually documented as: start:NA:end • “Compound” unitigs demarcate bubbles or other ambiguities in the graph • Edges grouped as: start:via:end • via edge indicates which branch of a fork to take • “Contained” unitigs are the paths within another compound unitig
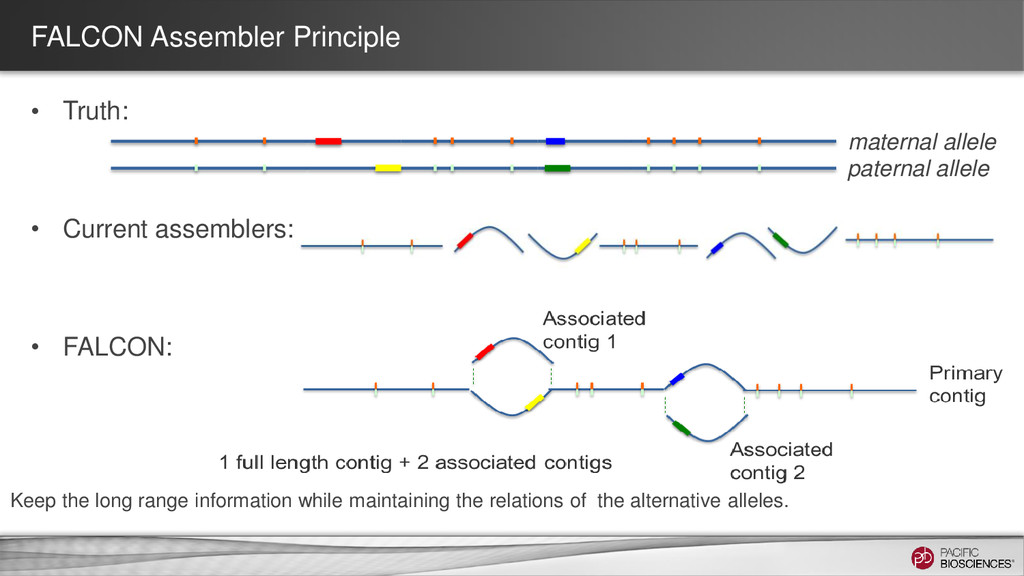
In CHM13 assembly, Contig 39 (blue) mapped to two different human chromosomes. The first 3 MB aligned with chr13 and the last 17 MB with chr14 • FALCON includes Python code to identify neighbors in the string graph, which represent alternative hypotheses for the assembly For the very adventurous: https://dl.dropboxusercontent.com/u/38943405/assembly_graph_notebook/asm_g raph_exploration_notebook_CHM13.slides.html
– Fix bugs • V 0.3 – New Dalinger version – Support for 100 kb subreads – New consensus code for superior diploid handling – Other features under discussion on FALCON’s Github page – Release TBD 20
Pacific Biosciences, the Pacific Biosciences logo, PacBio, SMRT, SMRTbell, and Iso-Seq are trademarks of Pacific Biosciences. BluePippin and SageELF are trademarks of Sage Science. NGS-go and NGSengine are trademarks of GenDx. All other trademarks are the sole property of their respective owners.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Preparing the FALCON Configuration File [General] job_type=local • Alternative is](https://files.speakerdeck.com/presentations/03a9719f12ee4351a87867beff0e89ab/slide_6.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}