of California, Inc. All rights reserved. For Research Use Only. Not for use in diagnostic procedures.. Nicole Rapicavoli Field Applications Scientist October 2014 Barcoding for Amplicon Sequencing Using SMRT® Analysis V2.3
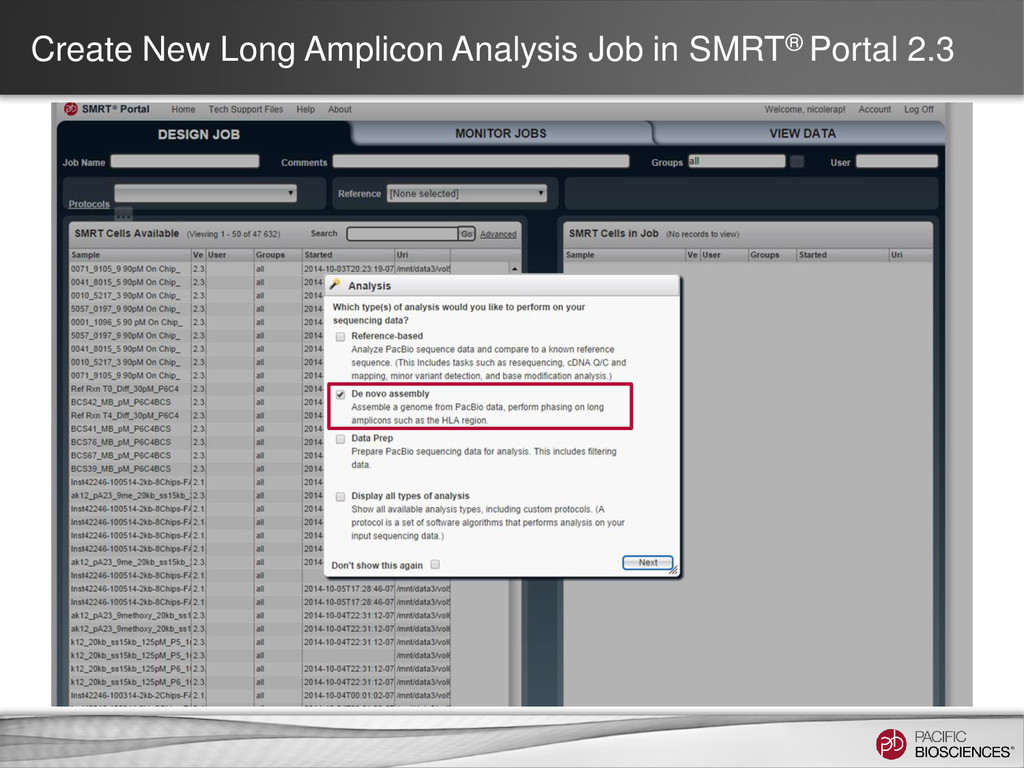
full-length sequencing. After the training, you will be able to • Be able to choose the best multiplexing strategy for your experimental design. • Understand the Long Amplicon Analysis method. • Run a Long Amplicon Analysis job and understand the reports generated in SMRT® Portal. • SMRT® Technology • PacBio® System Workflow • General Understanding of SMRT Portal
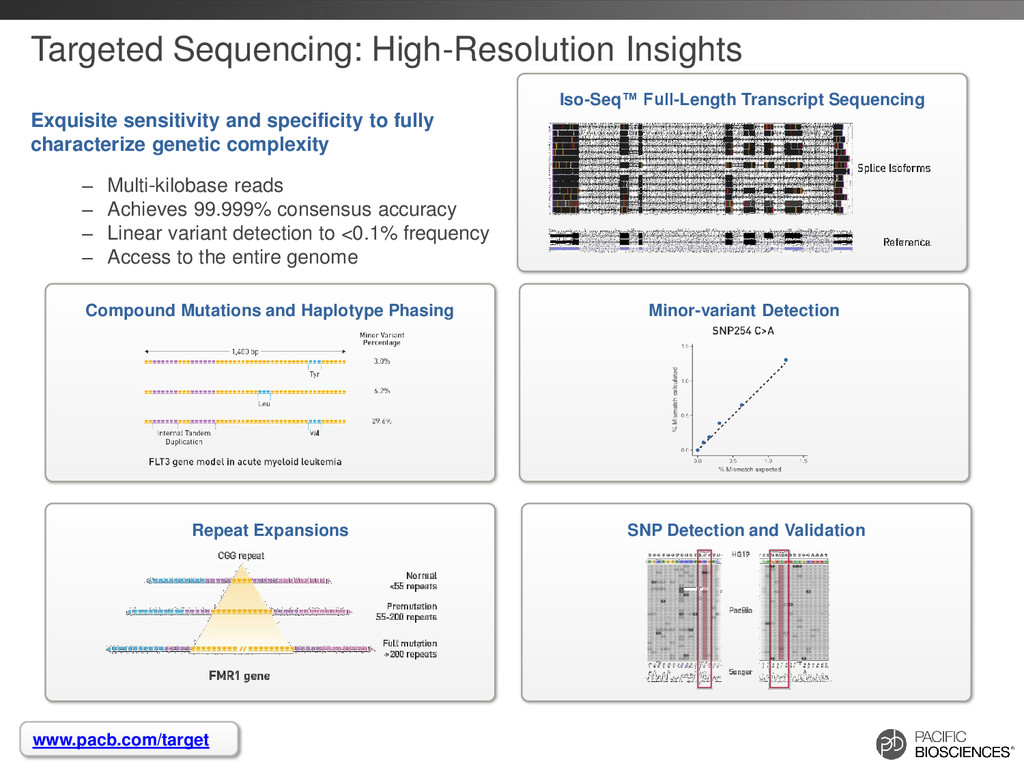
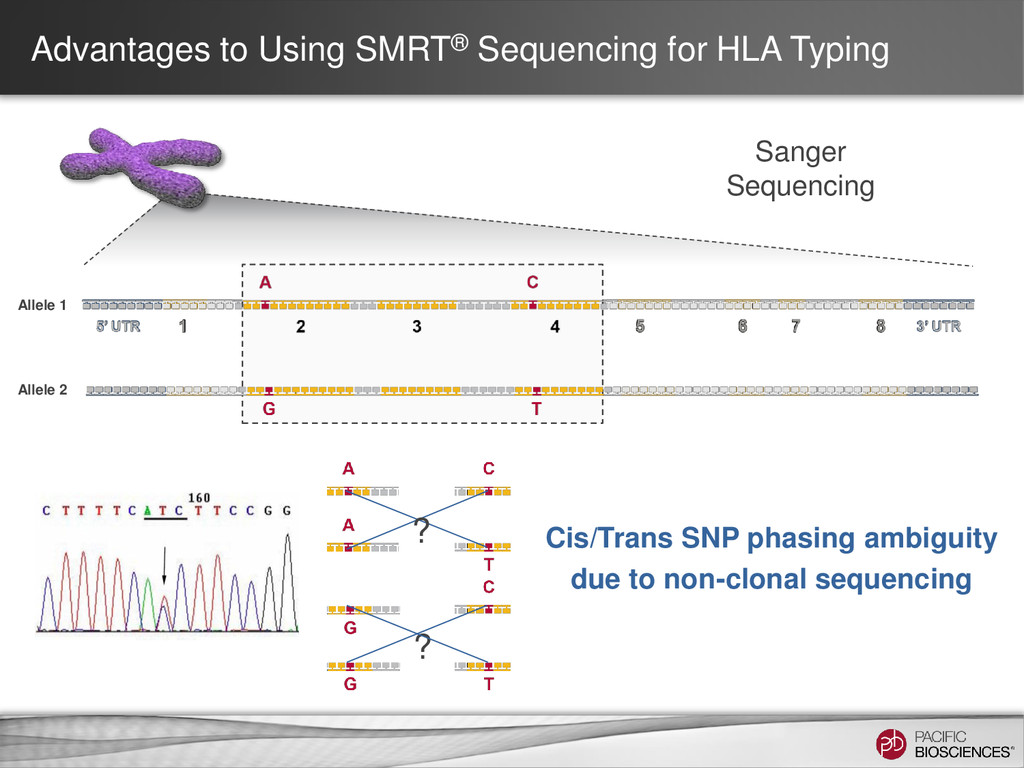
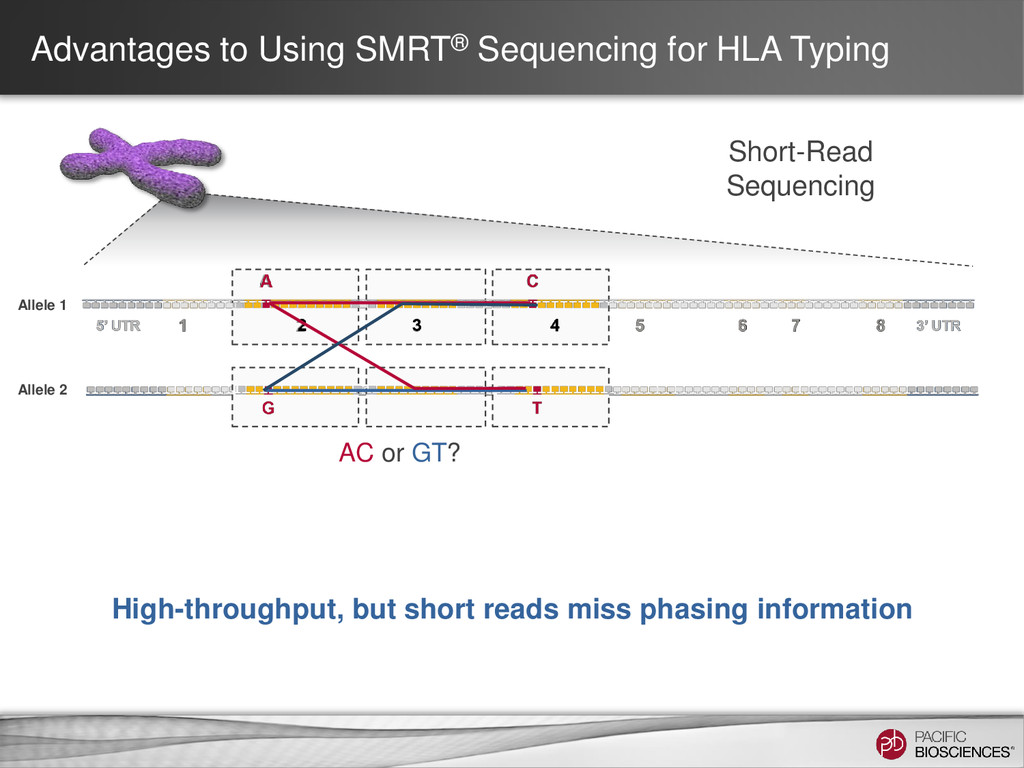
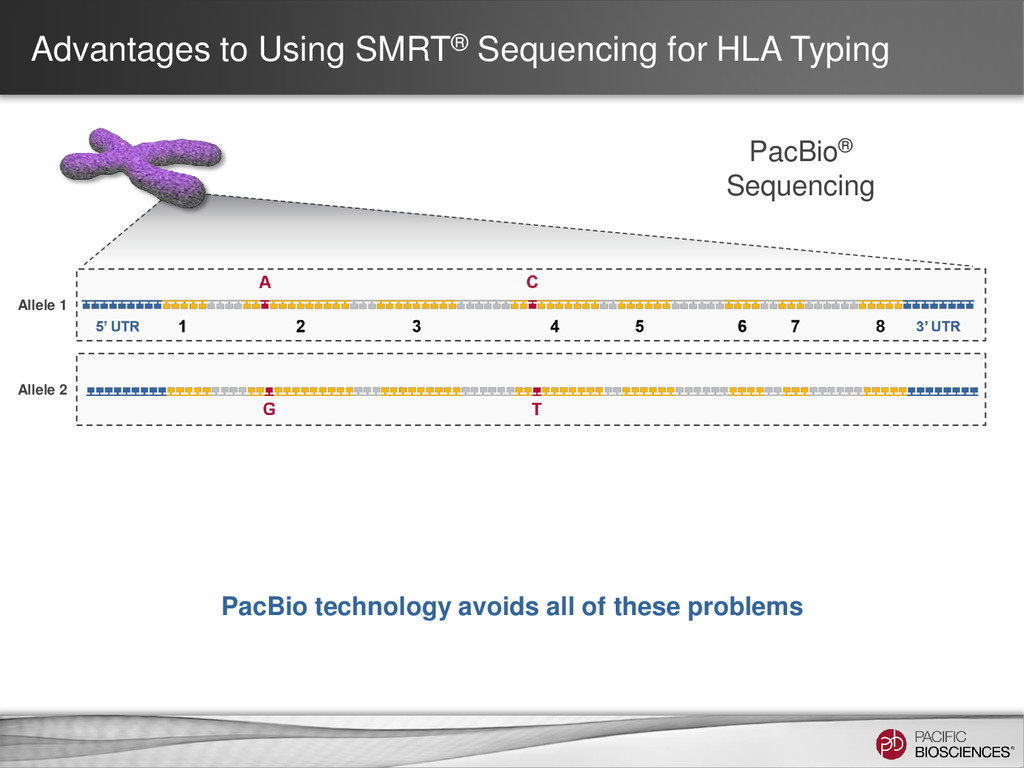
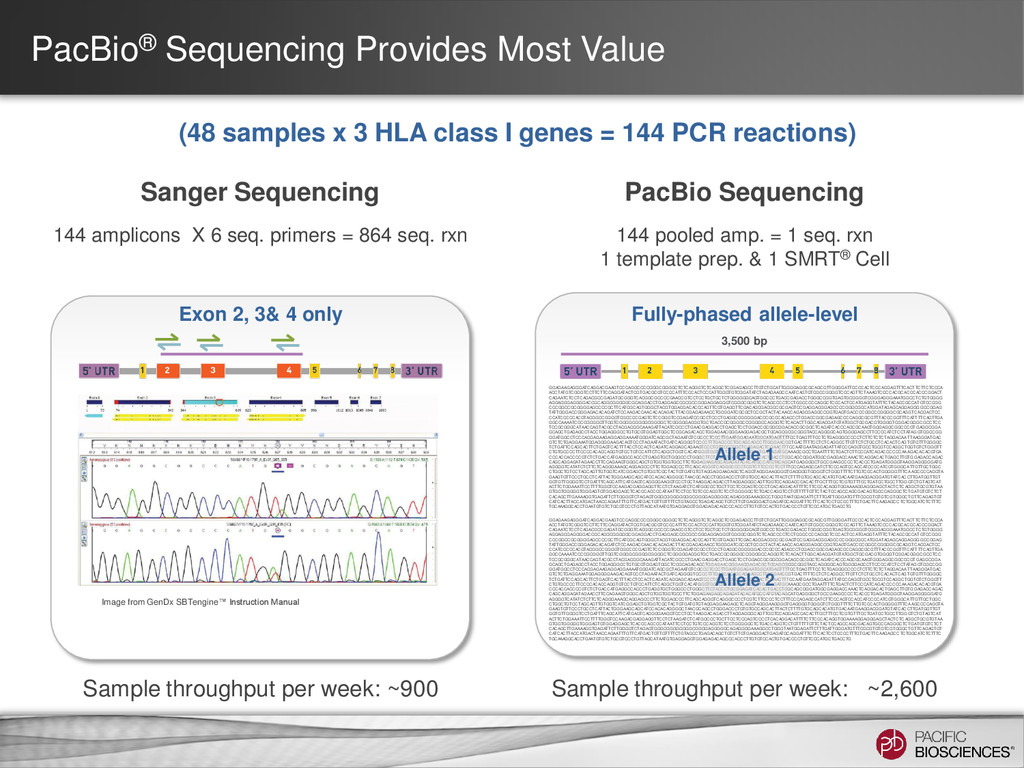
Sequencing can achieve greater than 99.999% (QV 50) accurate sequencing results for targeted sequencing applications – Near-perfect consensus accuracy – Best coverage uniformity with no amplification and minimal GC bias – Improved mappability with longer average read lengths • Flexibility in amplicon size allow for customized solutions for complex SNP detection applications • Sequencing of long amplicons with multi-kilobase read lengths provides direct strand-specific haplotype phasing • Single-molecule sequencing detects low-frequency minor variants at high- resolution 4
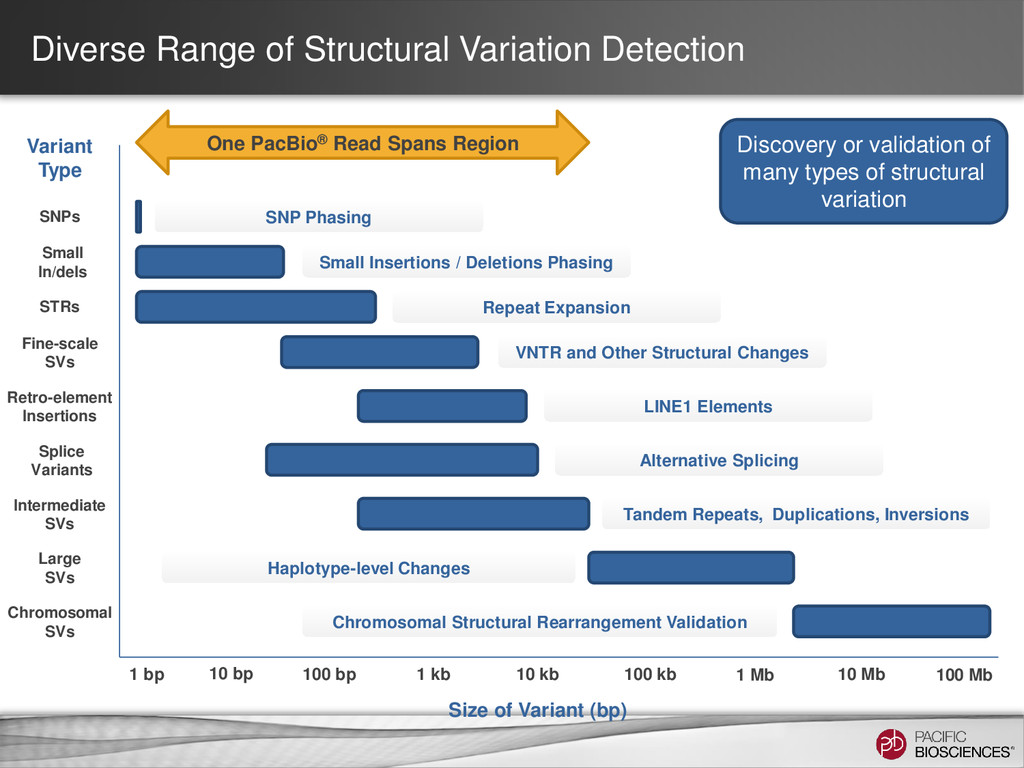
100 bp 1 kb 10 kb 100 kb 1 Mb 10 Mb 100 Mb Size of Variant (bp) Variant Type SNPs SNP Phasing Small In/dels Small Insertions / Deletions Phasing STRs Repeat Expansion Fine-scale SVs VNTR and Other Structural Changes Retro-element Insertions LINE1 Elements Splice Variants Alternative Splicing Intermediate SVs Tandem Repeats, Duplications, Inversions Large SVs Haplotype-level Changes Chromosomal SVs Chromosomal Structural Rearrangement Validation One PacBio® Read Spans Region Discovery or validation of many types of structural variation
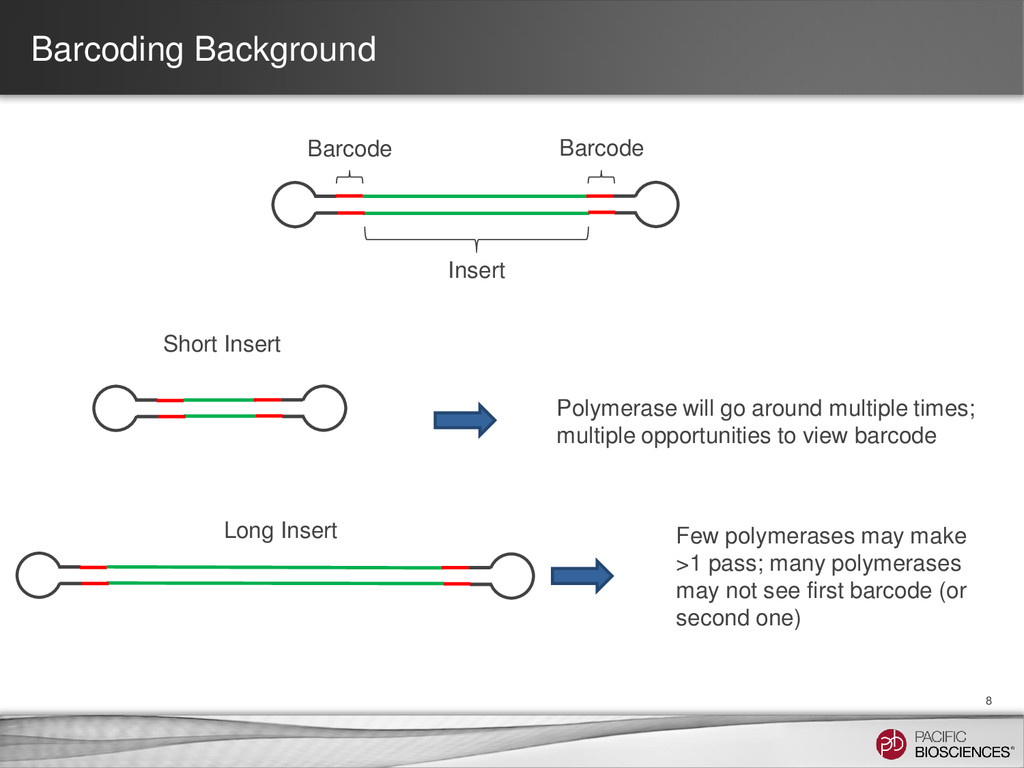
go around multiple times; multiple opportunities to view barcode Long Insert Few polymerases may make >1 pass; many polymerases may not see first barcode (or second one)
the same on both sides of the insert. – Recommended for inserts longer than 3 kb. • Paired Mode: (aka Paired/Asymmetric) – Different barcode sequences on either end of the insert. – Only recommended for sequences shorter than 3 kb. – Not yet validated. 9
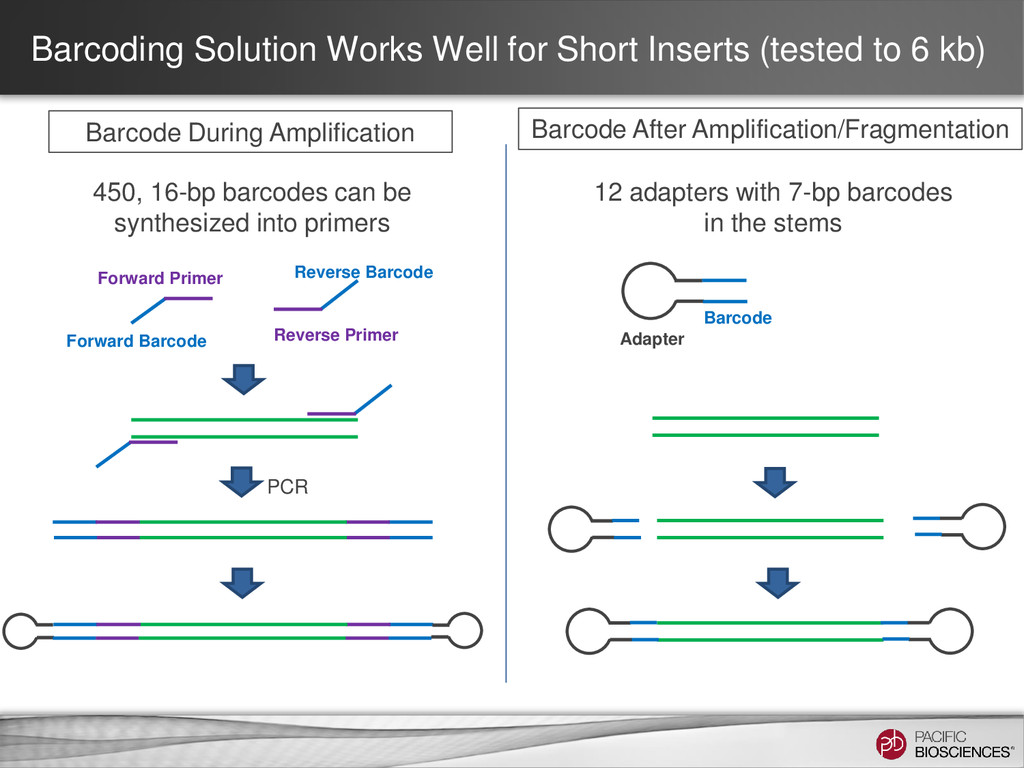
kb) Barcode During Amplification Barcode After Amplification/Fragmentation 450, 16-bp barcodes can be synthesized into primers Forward Primer Forward Barcode Reverse Primer Reverse Barcode 12 adapters with 7-bp barcodes in the stems Barcode Adapter PCR
<5 kb Inserts ≥5 kb Barcoding During Amplification (Primers) • Amplicon Panels • HLA Class I Genes • Clone Validation • Targeted Viral • HLA Class II Genes • Clone Validation • Full-length HIV • Structural Variants Barcoding After Amplification or Fragmentation (Adapters) • Amplicon Panels • Targeted Viral • Not recommended. • Instead pool non- overlapping and assemble in HGAP: BACs Fosmids Key Questions • How do you incorporate the barcode? • How do you optimize the number of barcodes you see?
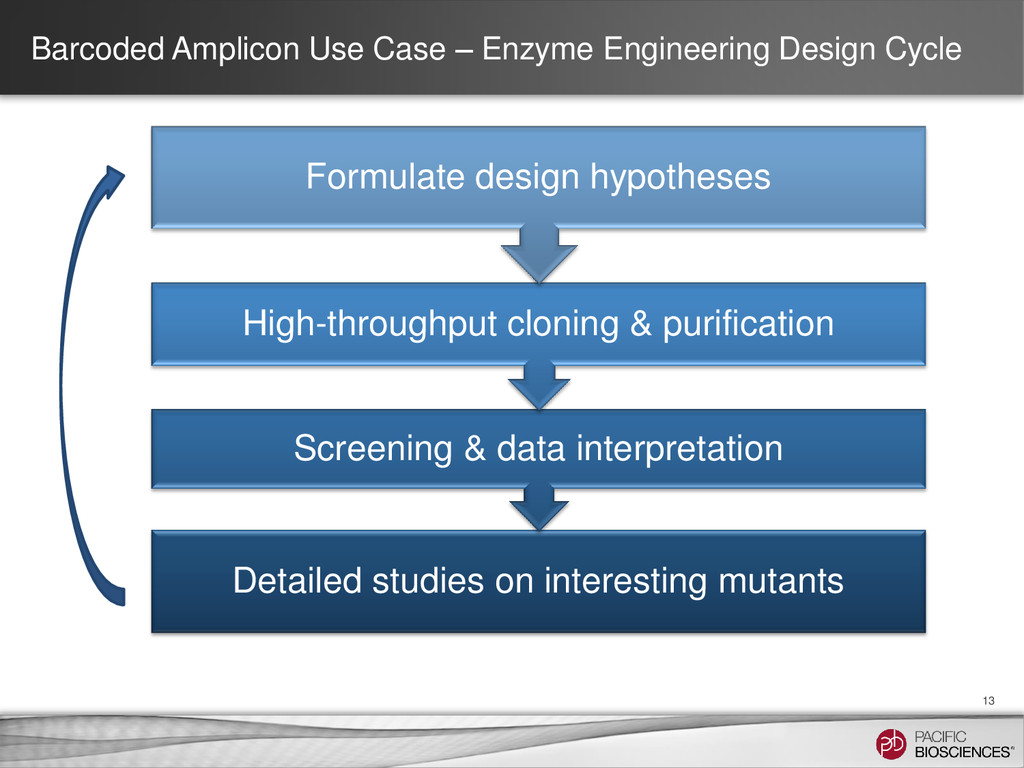
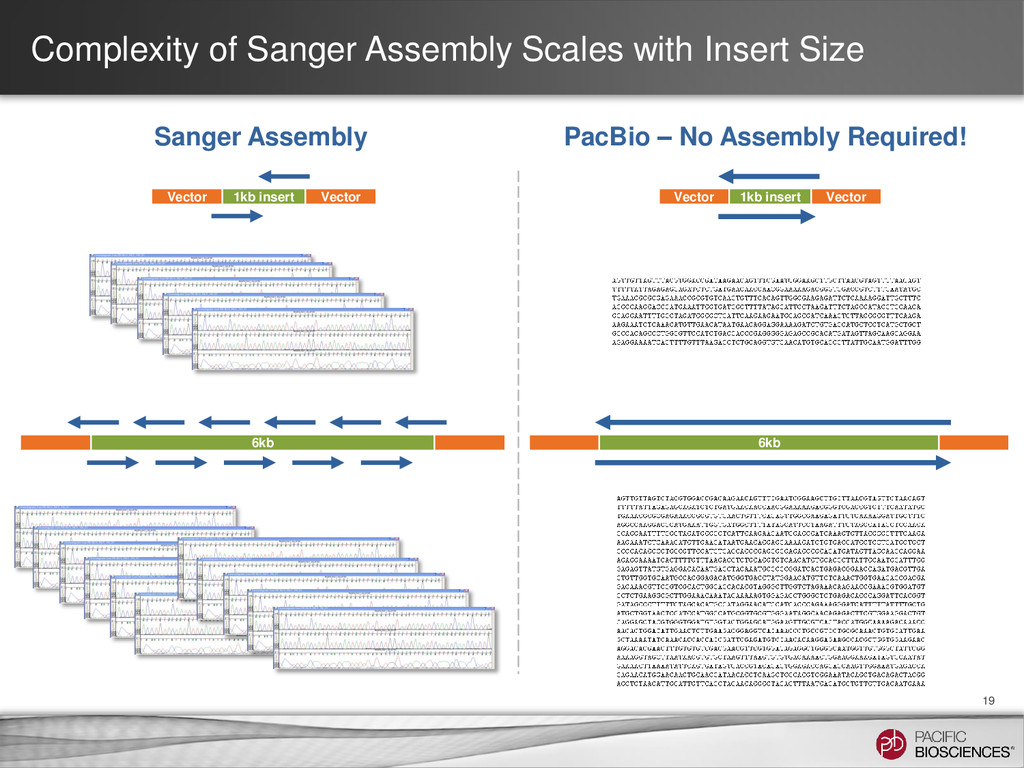
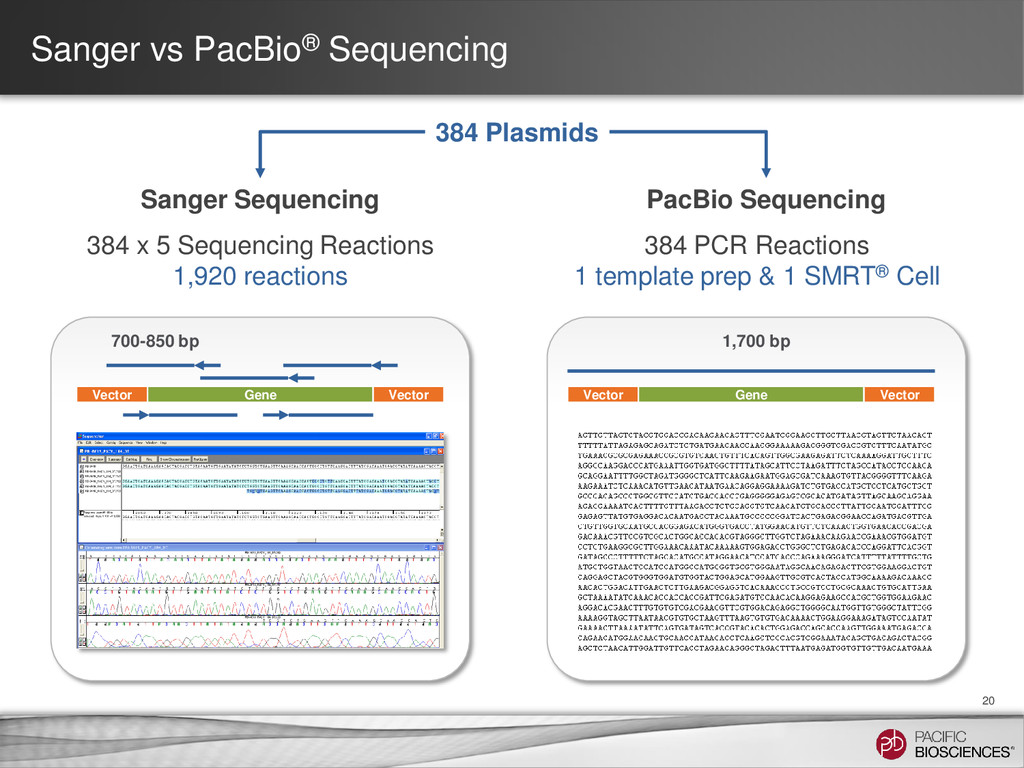
14 Goal • To compare a Sanger workflow with a PacBio® workflow (including barcoding) for clone validation Scope • Validating 384 distinct clones, each 1.7 kb in length, with high sequence homology (~99%) Project Design • Sanger: Five amplicons of ~750 bp for each clone • PacBio: One amplicon of 1.7 kb for each clone Results • Sequenced all clones with 100% accuracy using one barcoded library and one SMRT® Cell
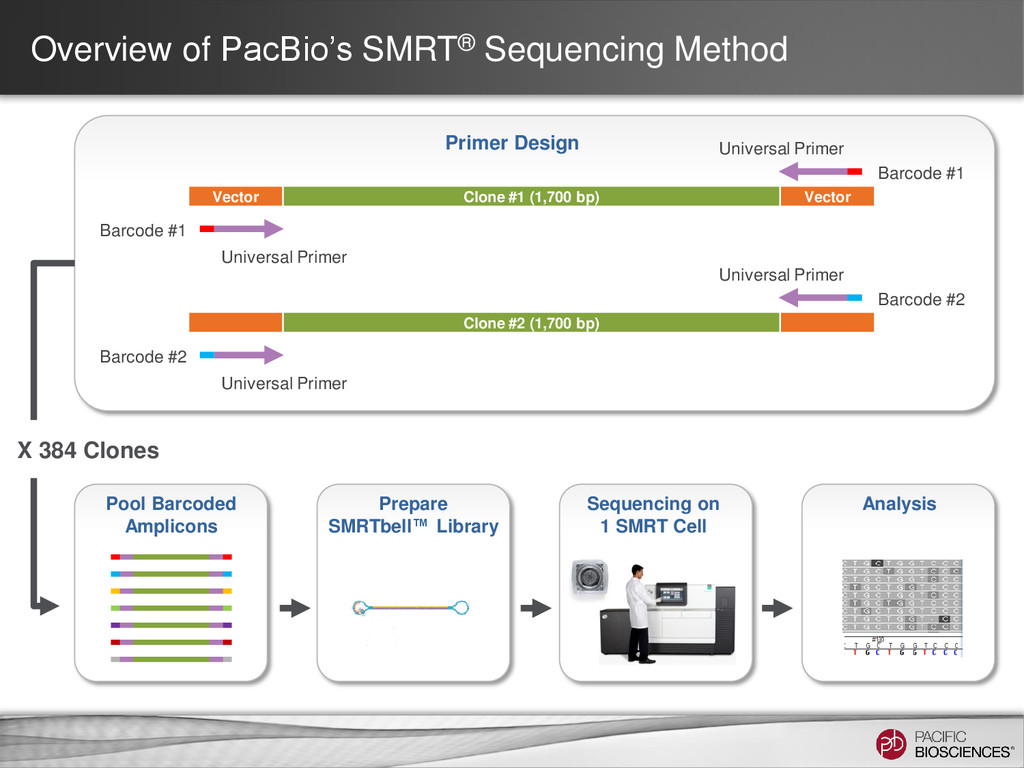
SMRTbell™ Library Sequencing on 1 SMRT Cell Analysis Primer Design Vector Clone #1 (1,700 bp) Vector Universal Primer Barcode #1 Universal Primer Barcode #1 Clone #2 (1,700 bp) Universal Primer Barcode #2 Universal Primer Barcode #2 X 384 Clones
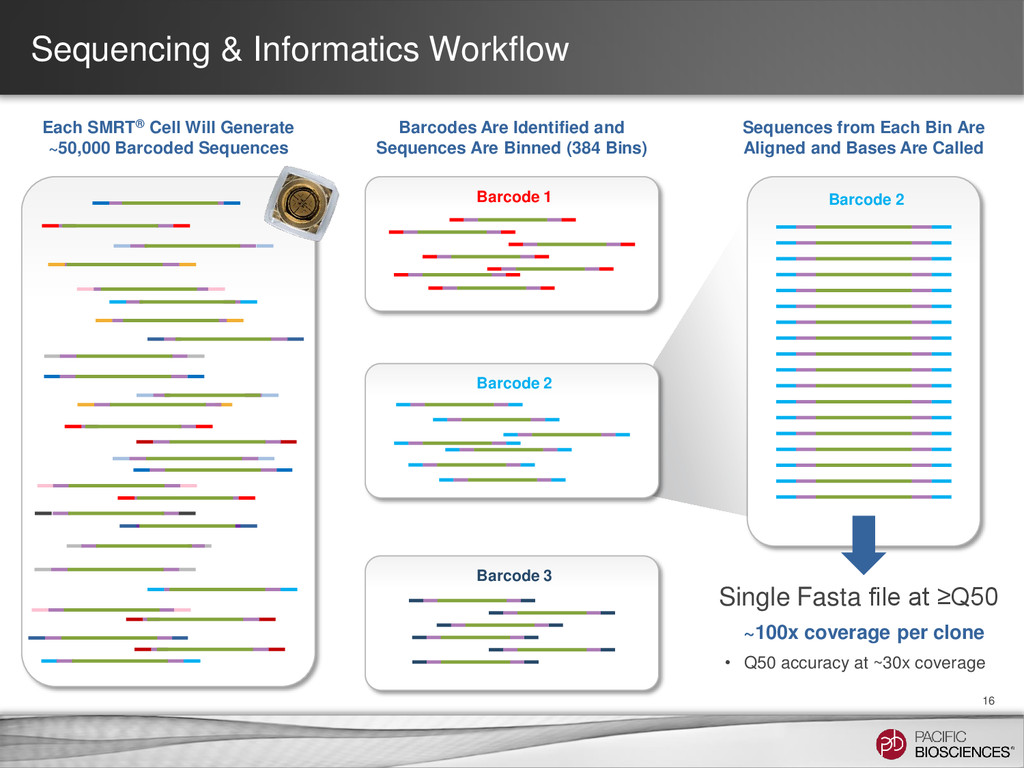
~50,000 Barcoded Sequences Barcode 2 Sequences from Each Bin Are Aligned and Bases Are Called ~100x coverage per clone • Q50 accuracy at ~30x coverage Single Fasta file at ≥Q50 Barcode 3 Barcode 1 Barcodes Are Identified and Sequences Are Binned (384 Bins) Barcode 2
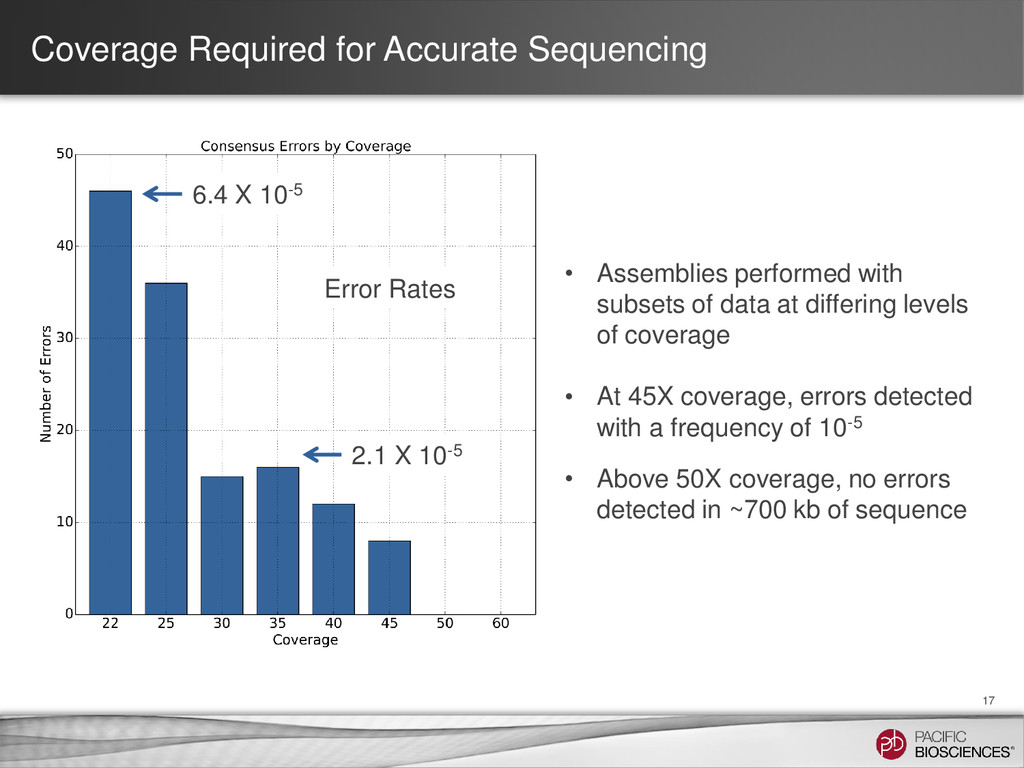
subsets of data at differing levels of coverage • At 45X coverage, errors detected with a frequency of 10-5 • Above 50X coverage, no errors detected in ~700 kb of sequence 6.4 X 10-5 2.1 X 10-5 Error Rates
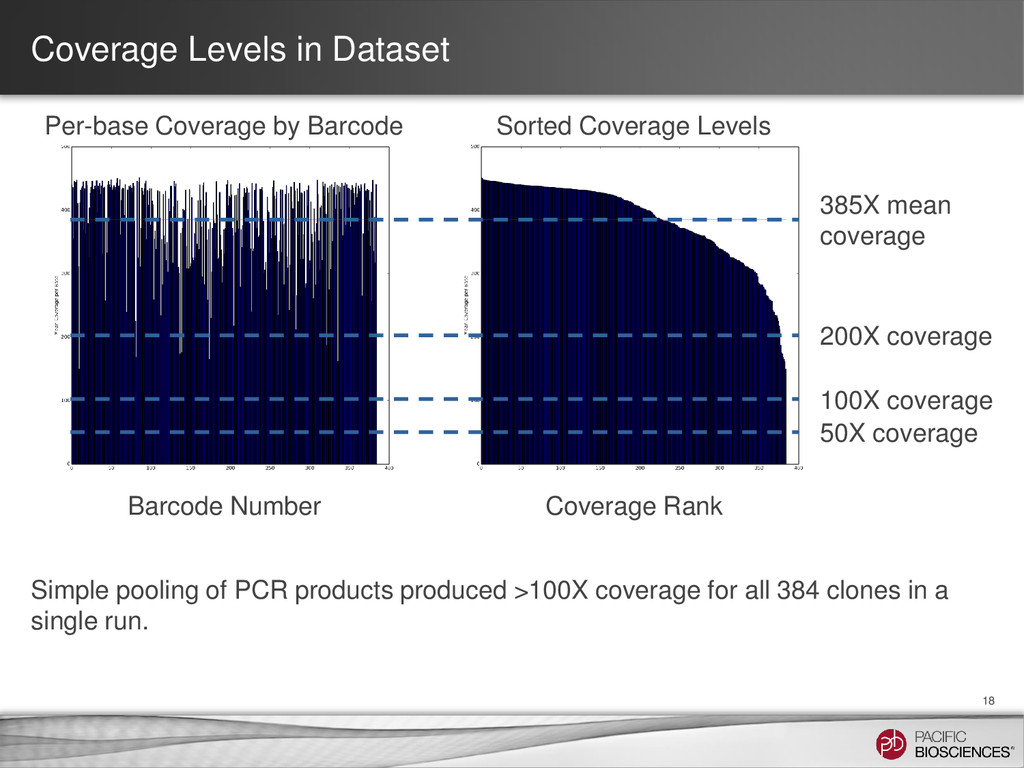
mean coverage Per-base Coverage by Barcode Barcode Number Coverage Rank Sorted Coverage Levels Simple pooling of PCR products produced >100X coverage for all 384 clones in a single run. 50X coverage
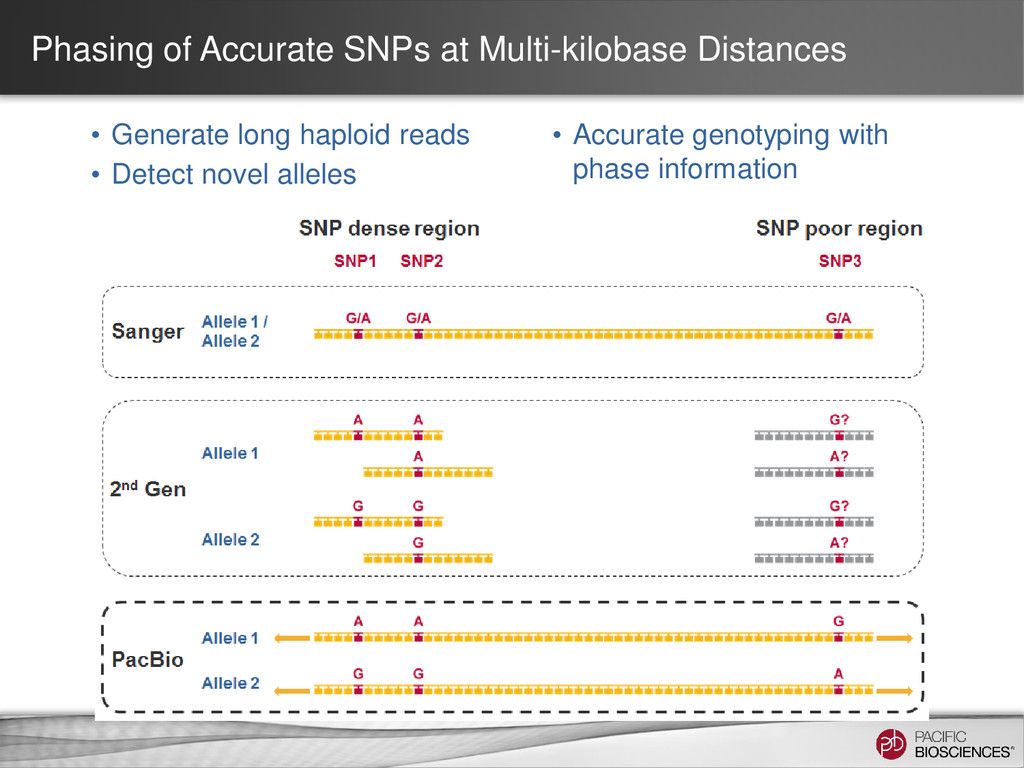
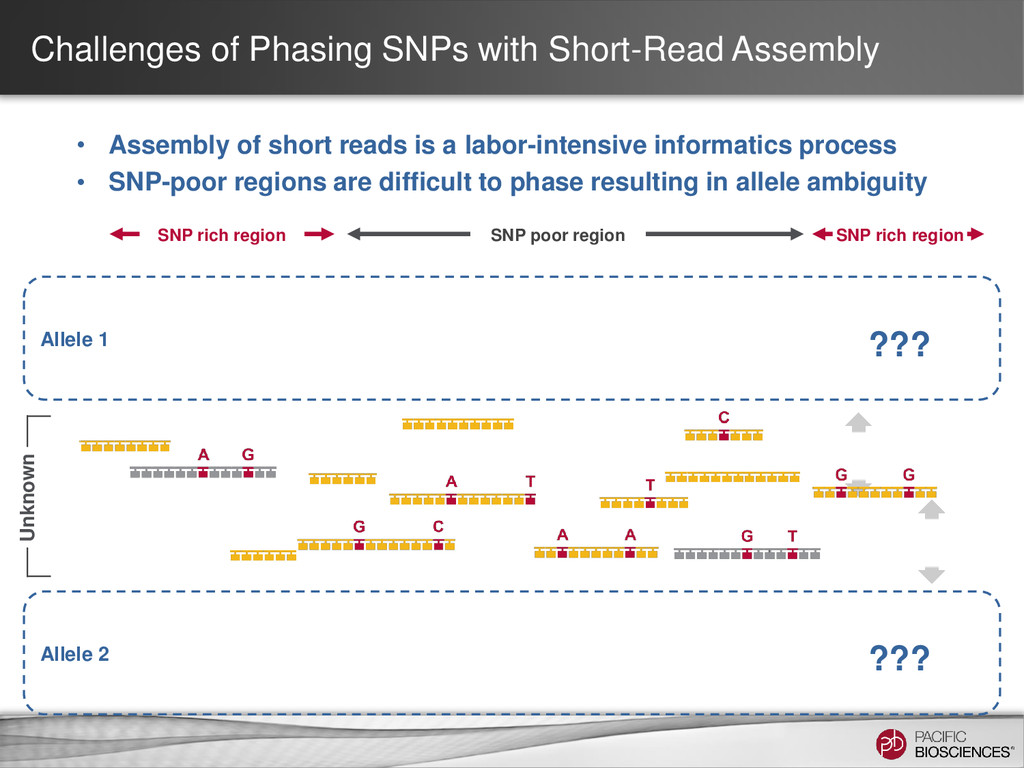
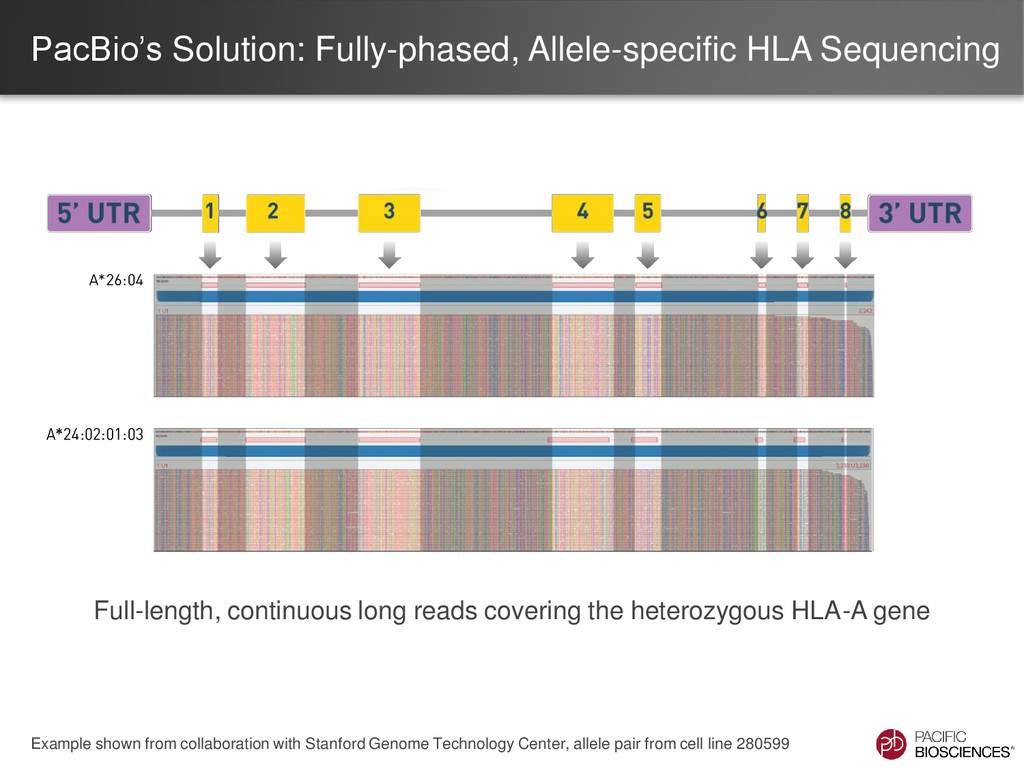
short reads is a labor-intensive informatics process • SNP-poor regions are difficult to phase resulting in allele ambiguity Allele 1 Allele 2 SNP rich region SNP poor region SNP rich region ??? ??? Unknown
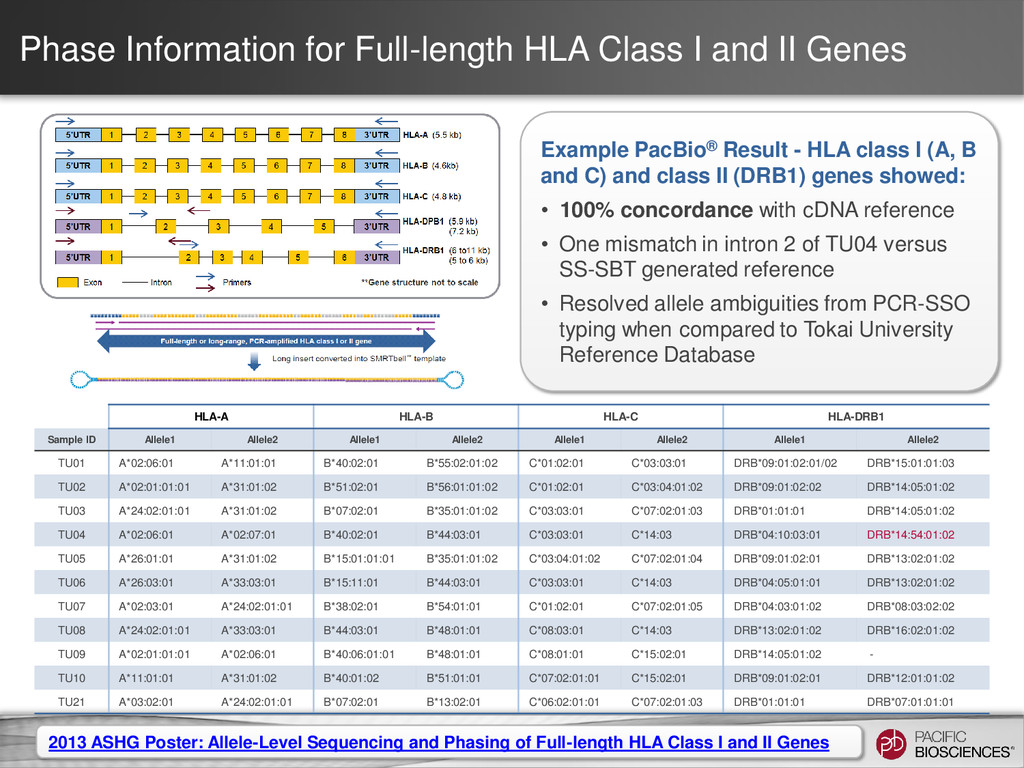
Four novel HLA alleles were identified using PacBio sequence data and have subsequently been submitted to the IMGT/HLA database: – A*68:01:02:02 – B*52:01:01:03 – C*02:02:02:02 – C*08:02:01:02 • One HLA B allele was corrected in the IMGT/HLA database: – B*27:05:02 Upcoming presentation by Collaborator at EFI 2014
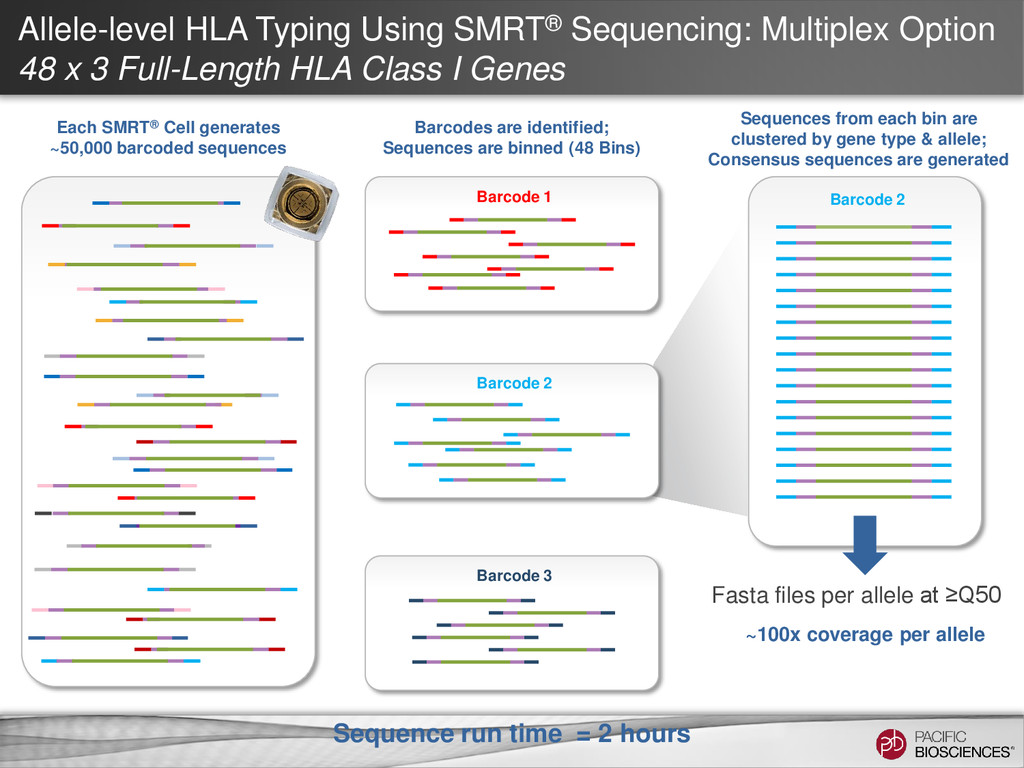
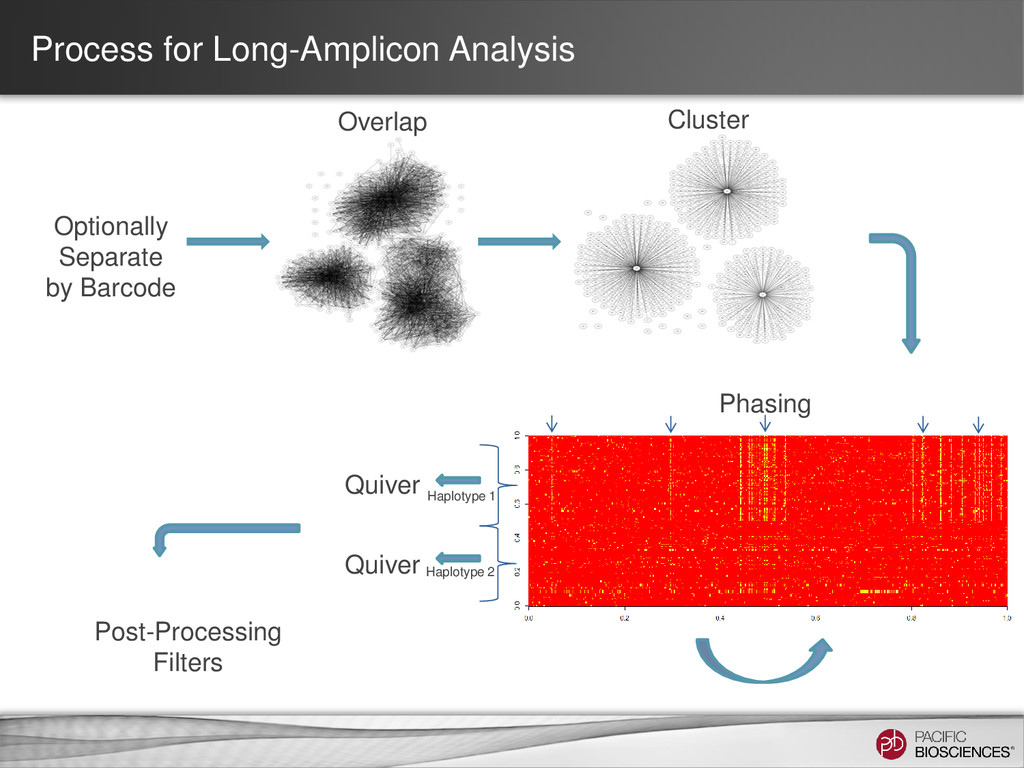
3 Full-Length HLA Class I Genes Each SMRT® Cell generates ~50,000 barcoded sequences Barcode 2 Sequences from each bin are clustered by gene type & allele; Consensus sequences are generated ~100x coverage per allele Fasta files per allele at ≥Q50 Barcode 3 Barcode 1 Barcodes are identified; Sequences are binned (48 Bins) Barcode 2 Sequence run time = 2 hours
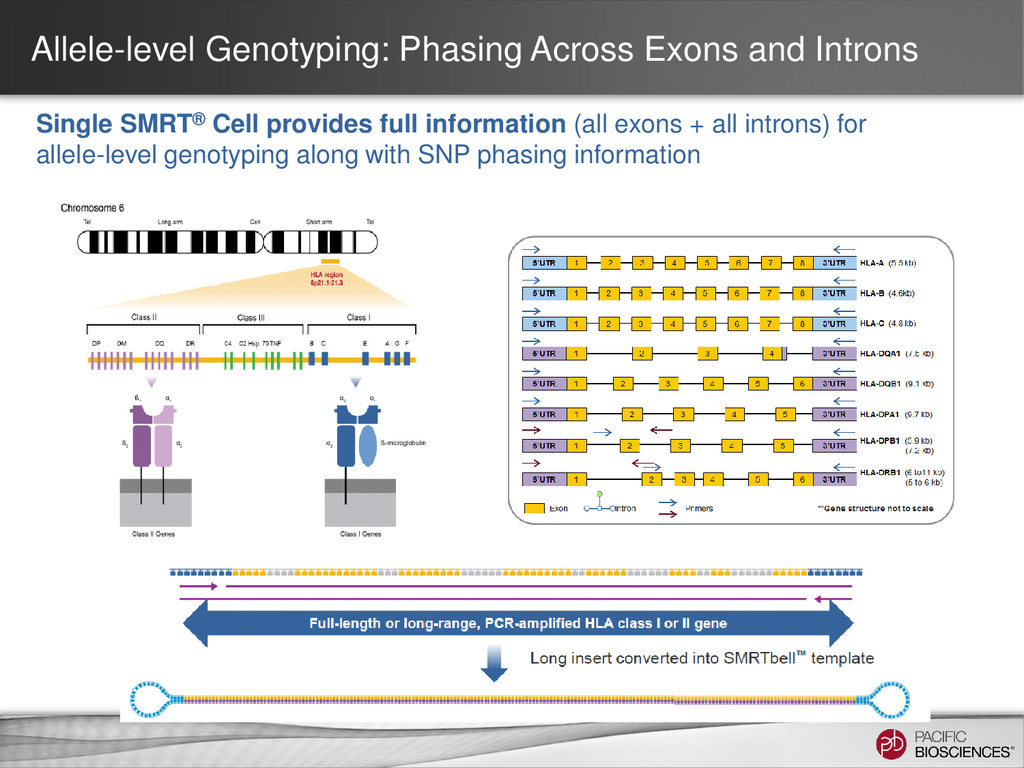
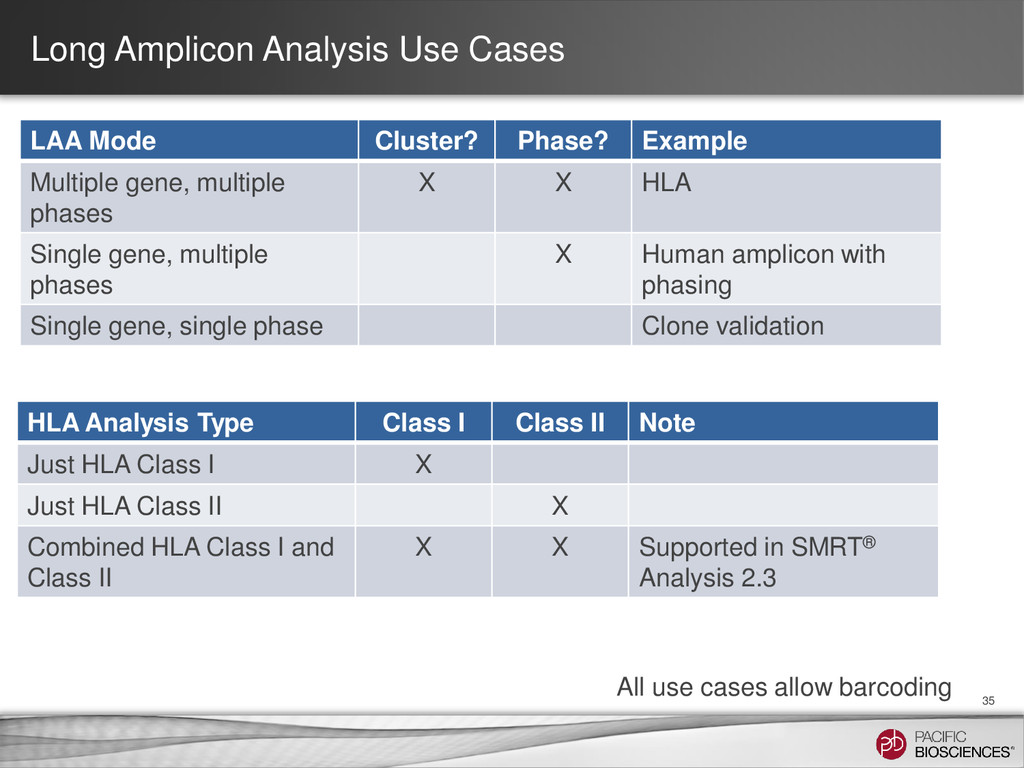
Multiple gene, multiple phases X X HLA Single gene, multiple phases X Human amplicon with phasing Single gene, single phase Clone validation 35 All use cases allow barcoding HLA Analysis Type Class I Class II Note Just HLA Class I X Just HLA Class II X Combined HLA Class I and Class II X X Supported in SMRT® Analysis 2.3
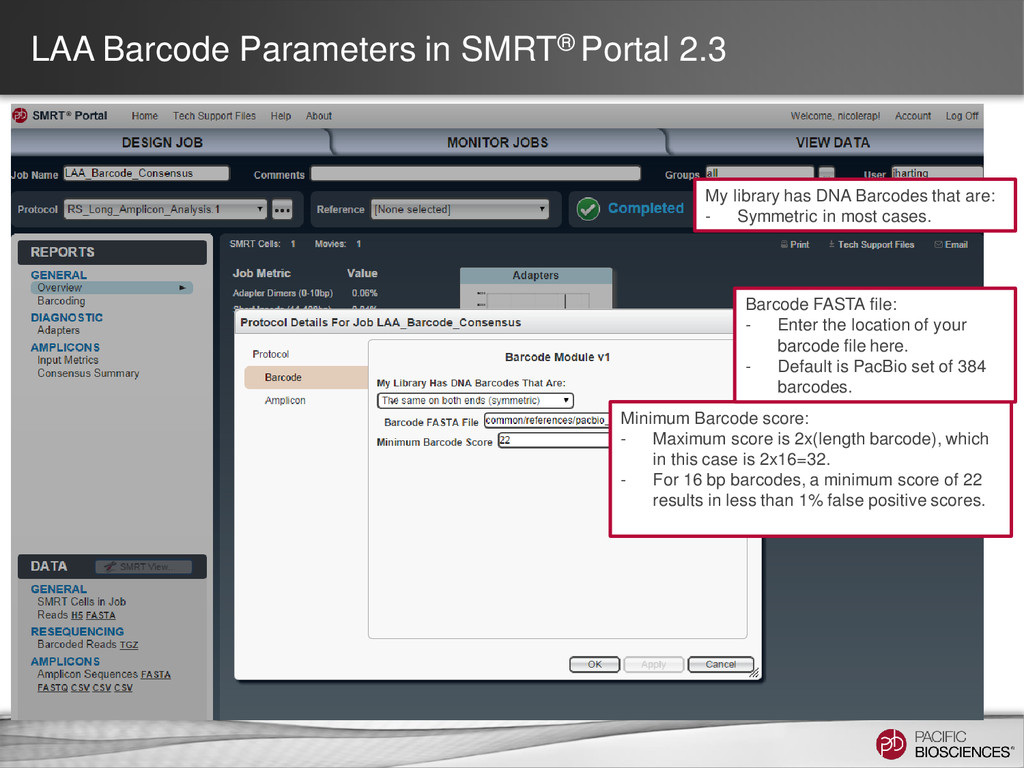
DNA Barcodes that are: - Symmetric in most cases. Minimum Barcode score: - Maximum score is 2x(length barcode), which in this case is 2x16=32. - For 16 bp barcodes, a minimum score of 22 results in less than 1% false positive scores. Barcode FASTA file: - Enter the location of your barcode file here. - Default is PacBio set of 384 barcodes.
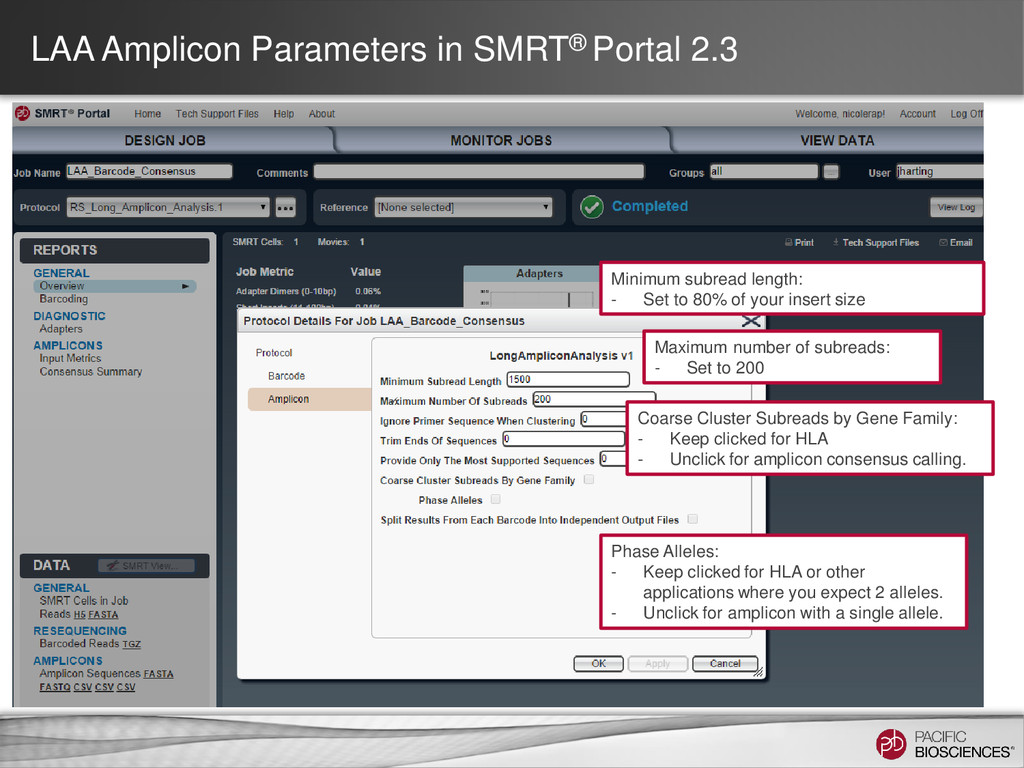
- Set to 80% of your insert size Coarse Cluster Subreads by Gene Family: - Keep clicked for HLA - Unclick for amplicon consensus calling. Maximum number of subreads: - Set to 200 Phase Alleles: - Keep clicked for HLA or other applications where you expect 2 alleles. - Unclick for amplicon with a single allele.
Pacific Biosciences, the Pacific Biosciences logo, PacBio, SMRT, SMRTbell, and Iso-Seq are trademarks of Pacific Biosciences in the United States and/or other countries. All other trademarks are the sole property of their respective owners.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}