of California, Inc. All rights reserved. For Research Use Only. Not for use in diagnostic procedures.. John Harting Bioinformatics Scientist, Applications Lab October 2014 Barcoding and Amplicon Sequencing Using SMRT® Analysis V2.3
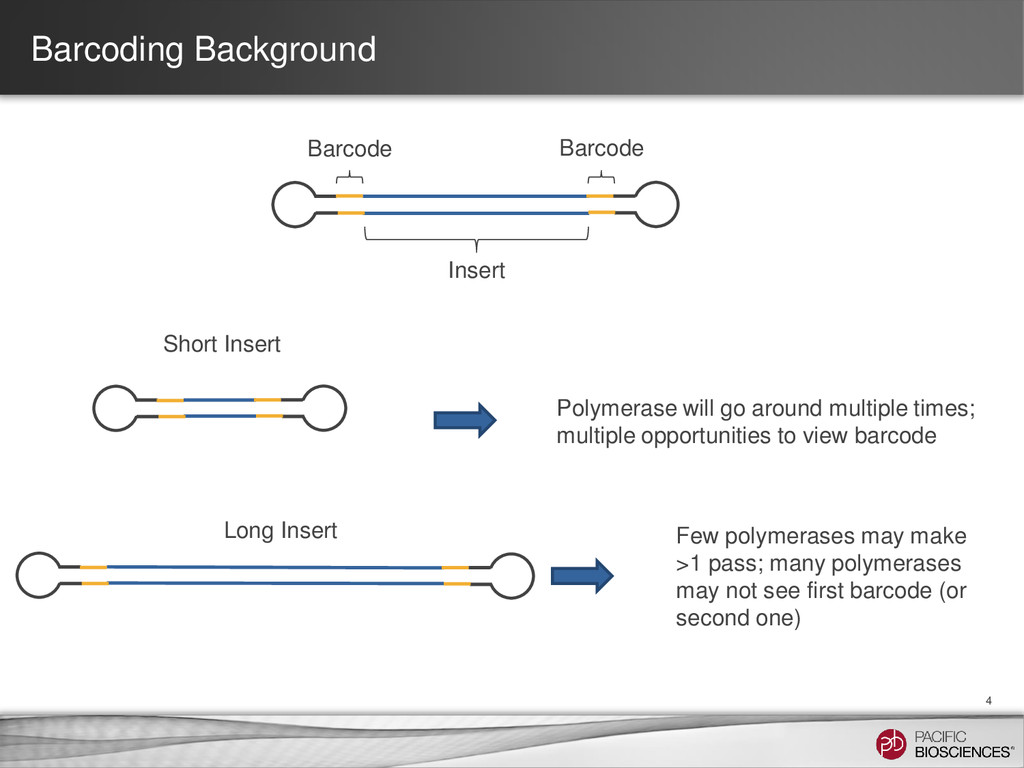
go around multiple times; multiple opportunities to view barcode Long Insert Few polymerases may make >1 pass; many polymerases may not see first barcode (or second one)
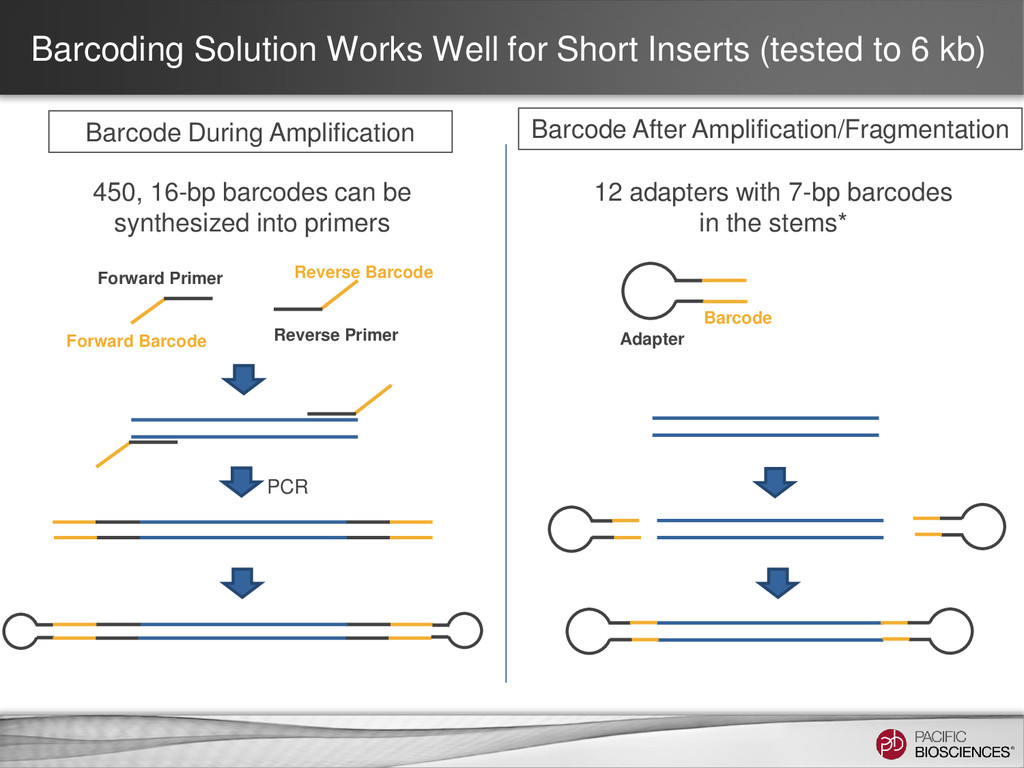
kb) Barcode During Amplification Barcode After Amplification/Fragmentation 450, 16-bp barcodes can be synthesized into primers Forward Primer Forward Barcode Reverse Primer Reverse Barcode 12 adapters with 7-bp barcodes in the stems* Barcode Adapter PCR
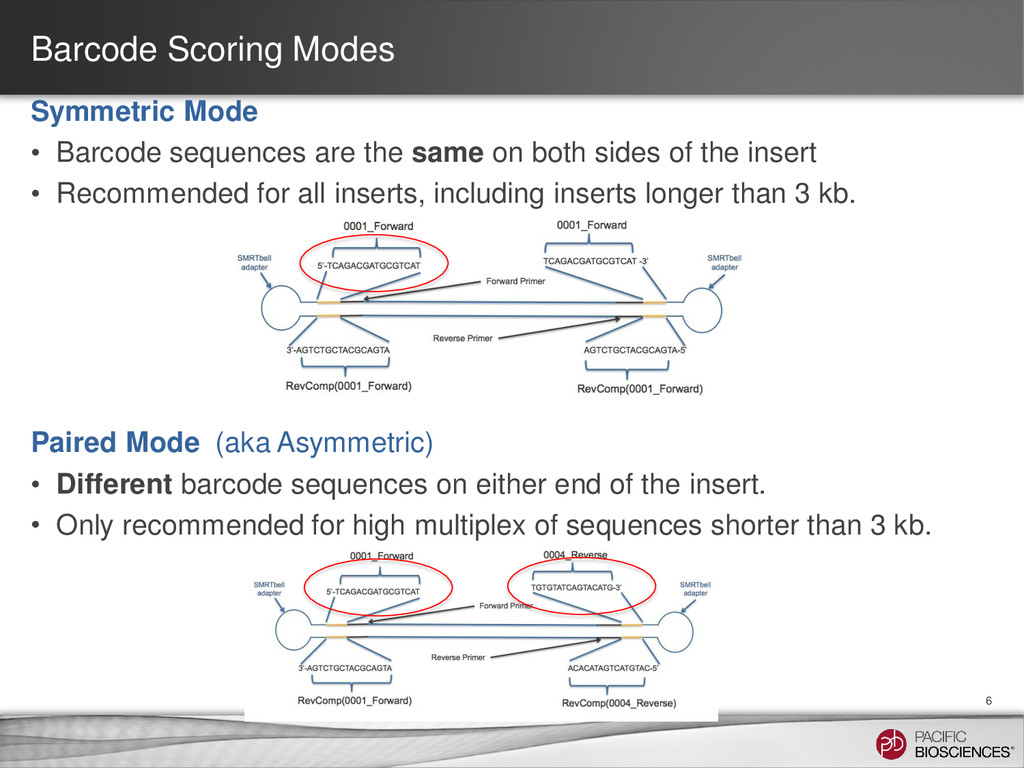
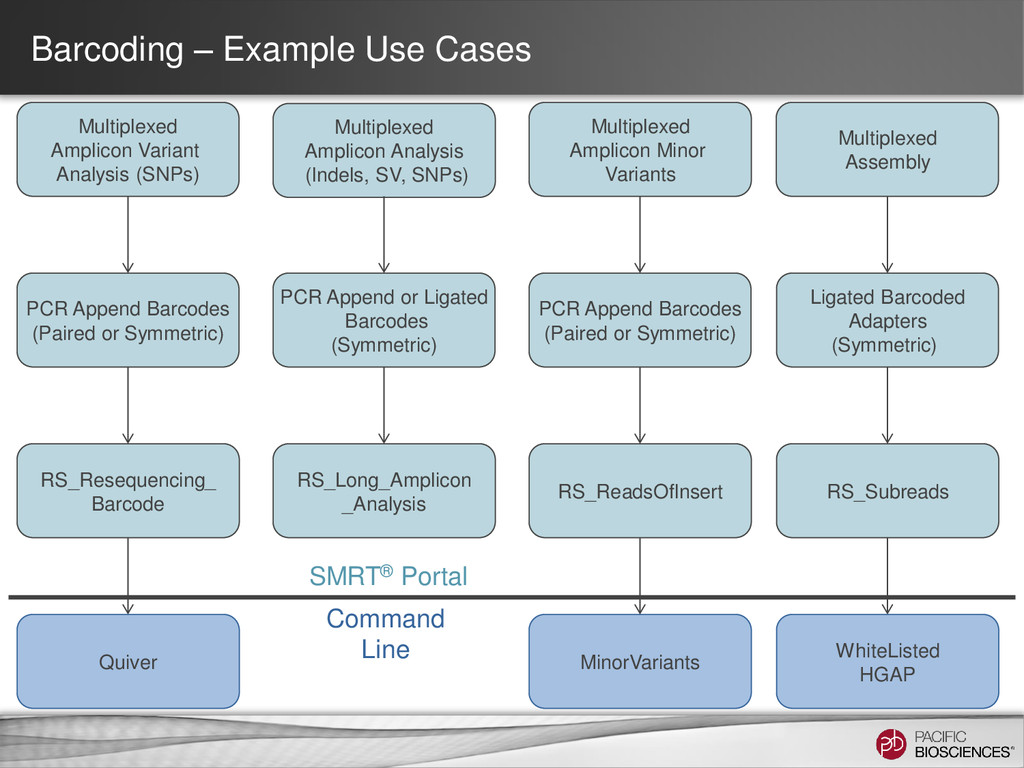
same on both sides of the insert • Recommended for all inserts, including inserts longer than 3 kb. Paired Mode (aka Asymmetric) • Different barcode sequences on either end of the insert. • Only recommended for high multiplex of sequences shorter than 3 kb. 6
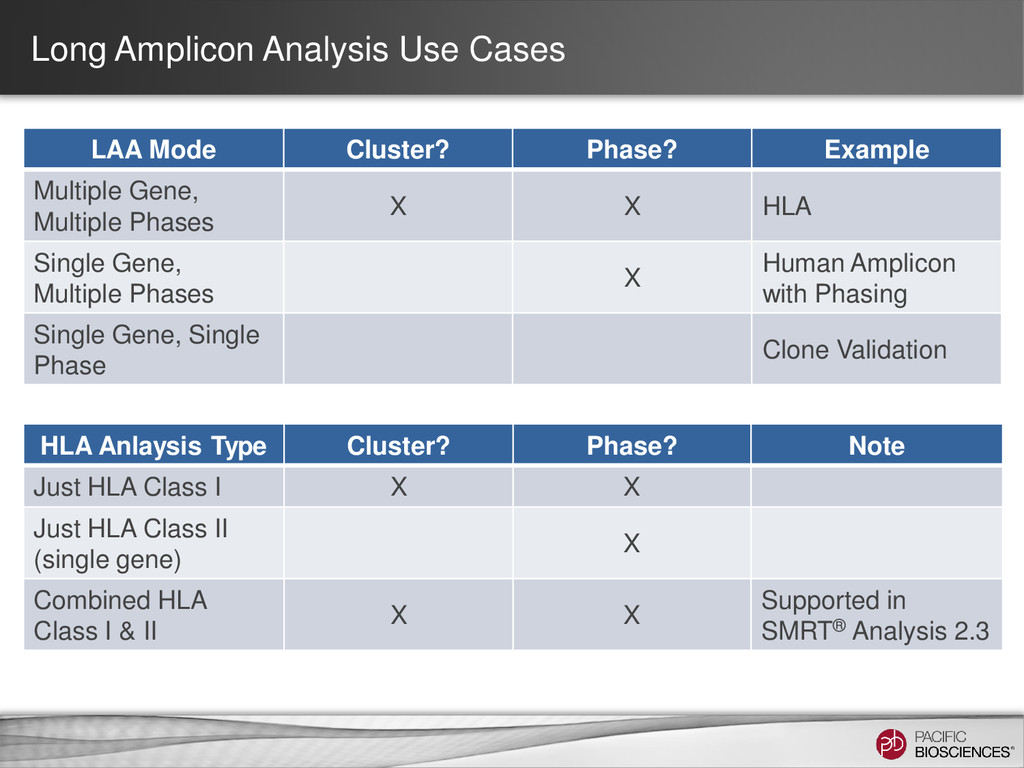
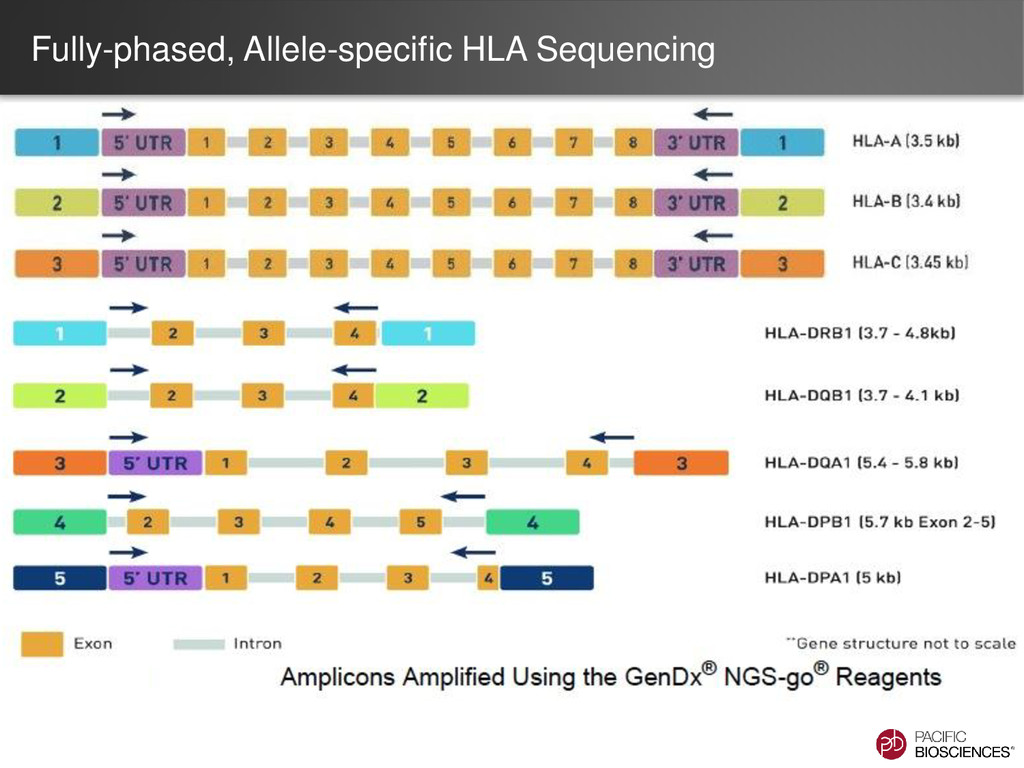
Multiple Gene, Multiple Phases X X HLA Single Gene, Multiple Phases X Human Amplicon with Phasing Single Gene, Single Phase Clone Validation HLA Anlaysis Type Cluster? Phase? Note Just HLA Class I X X Just HLA Class II (single gene) X Combined HLA Class I & II X X Supported in SMRT® Analysis 2.3
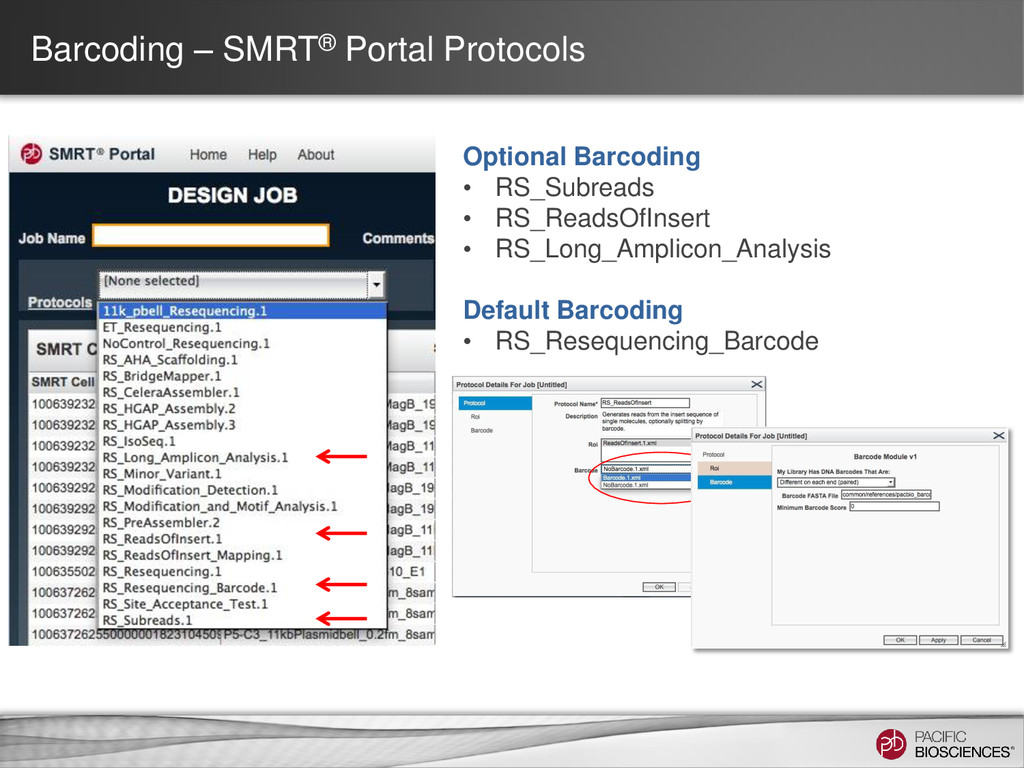
Portal • SMRT Pipe • Barcode Module • Whitelist Filter <param name="whiteList"> <value>/path/to/whitelist.txt</value> </param> • Command Line • barcodes - barcode fofn (from pbbarcode) • doBc - Specify a subset of all barcode • minBarcodeScore - Minimum average barcode • whiteList - A list of subreads to use in TXT or FASTA format.
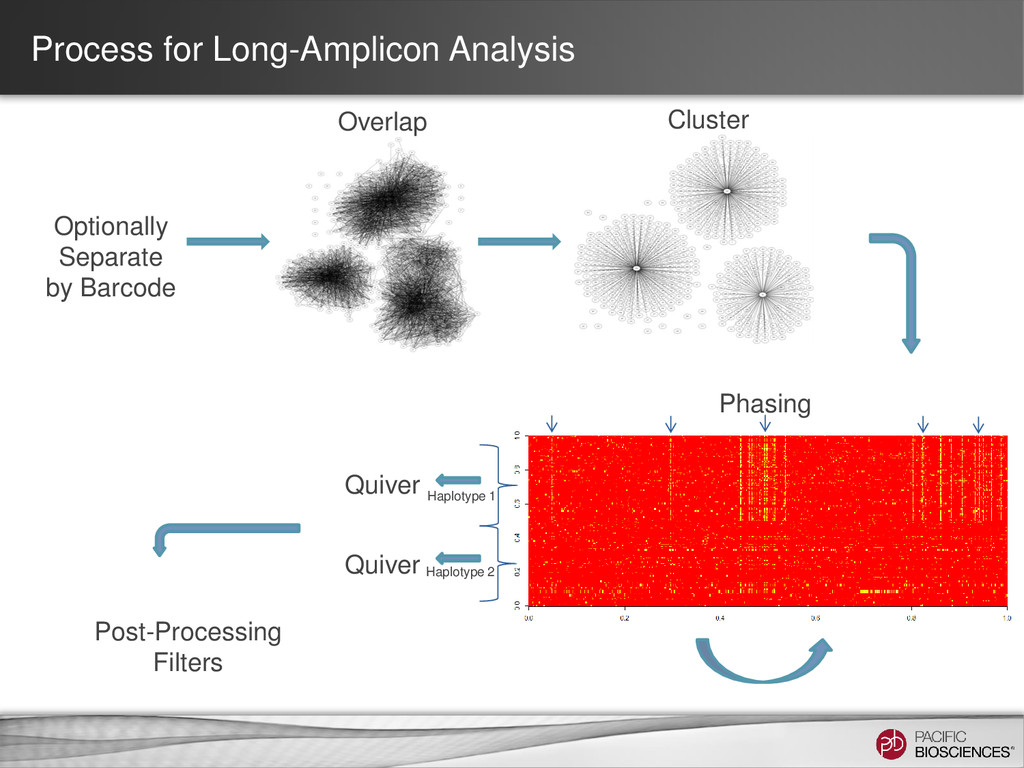
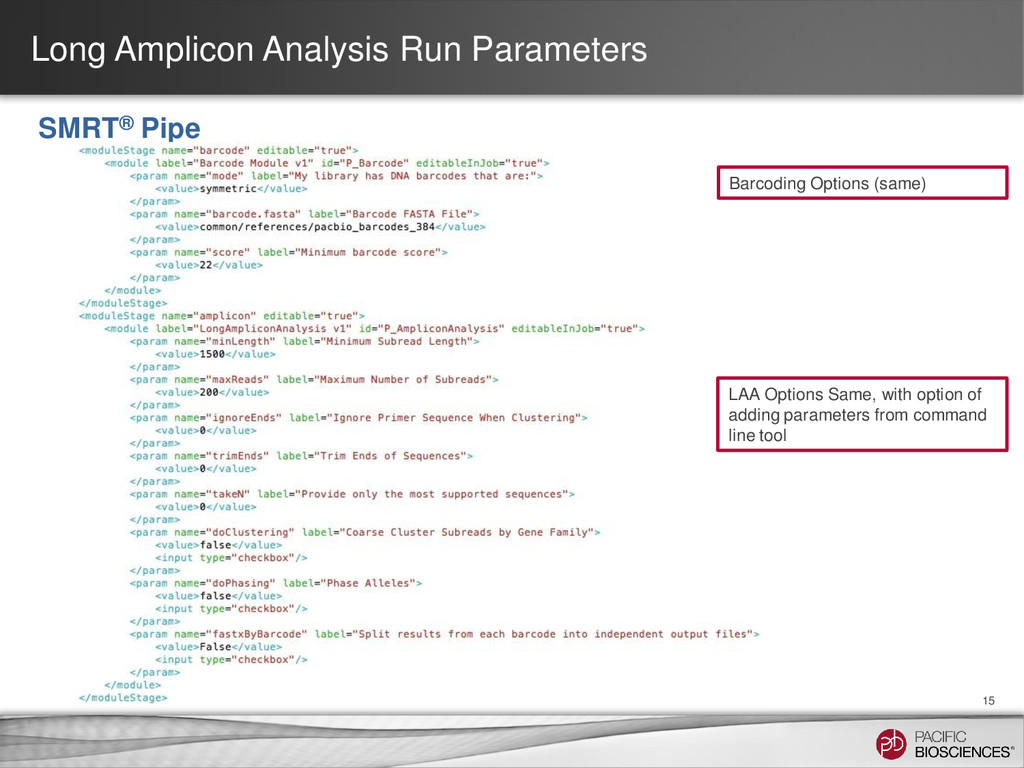
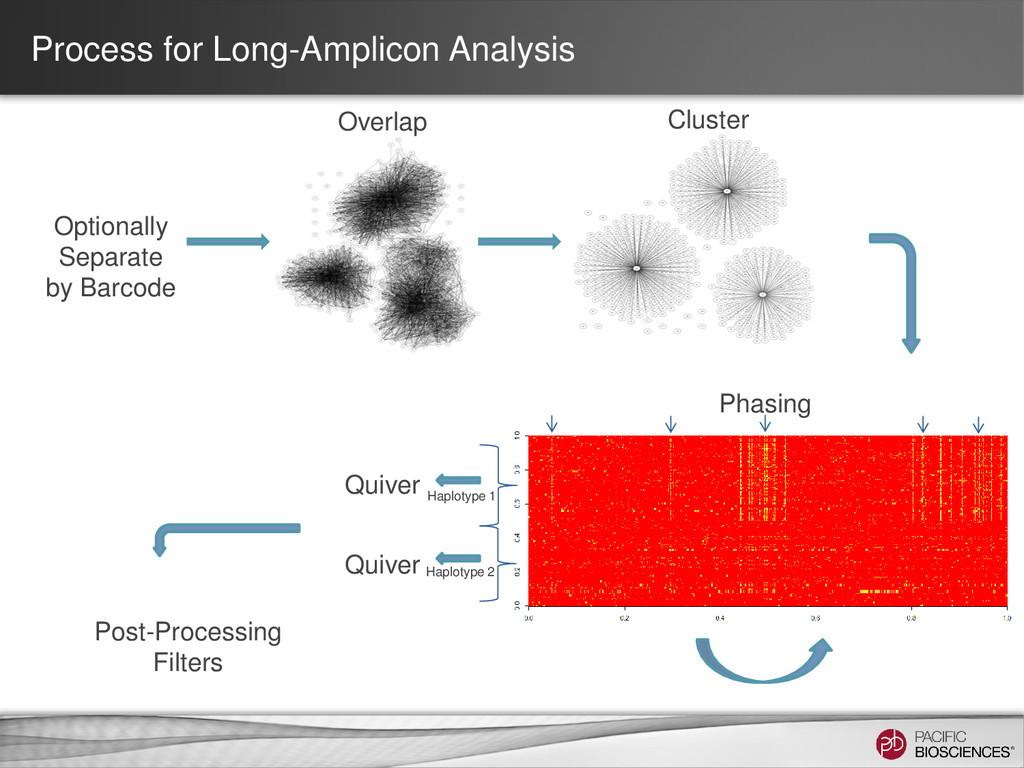
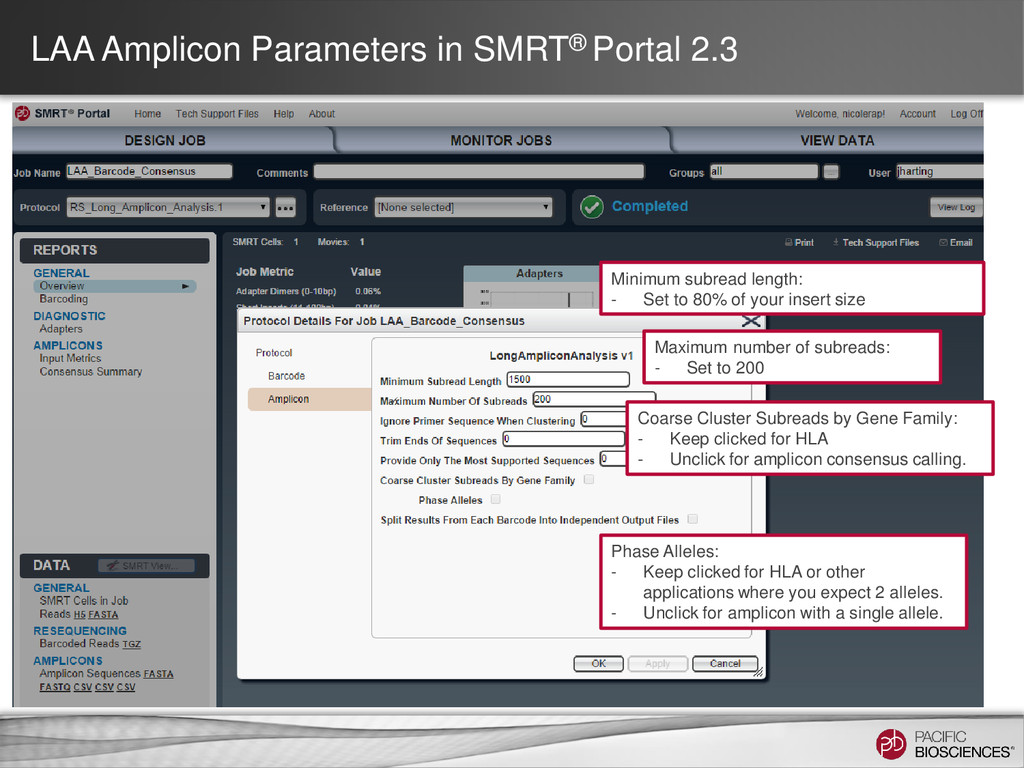
target amplicon* Coarse Cluster Subreads by Gene Family: - Keep clicked for HLA - Unclick for amplicon consensus calling. Maximum number of subreads: - Set to ~700 reads per Gene Phase Alleles: - Unclick for amplicons with a single allele. Long Amplicon Analysis Run Parameters 14 SMRT® Portal Ignore Primer When Clustering: - # bp to ignore on ends Trim Ends: - # bp to trim after consensus Split Results by Barcode: - Generate fasta/q file per barcode
• All the above plus lots more! • Extra Subread Criteria – maxLength, minReadScore, minSnr • Extra Clustering Criteria – maxClusters, clusterInflation (Markov) • Extra Phasing Criteria – minSplitScore, minSplitFraction, minSplitReads • Extra Filtering Options – minPredictedAccuracy, noChimeraFilter, chimeraScoreThreshold, convergenceFilter • Process Control – numThreads & forced threading across barcodes* *May cause out of memory error, only recommended for high memory machines
all filters Amplicon Analysis Summary (csv) • Pass/Fail status of filters for all consensus sequences Amplicon Analysis (csv) • Per-base coverage and QV scores Amplicon Analysis Zmws/Subreads (csv) • Mapping of zmws/subreads to consensus sequences Amplicon Analysis Chimeras Noise (fasta/q) • Consensus sequences failing filters. (Not available directly from SMRT Portal)
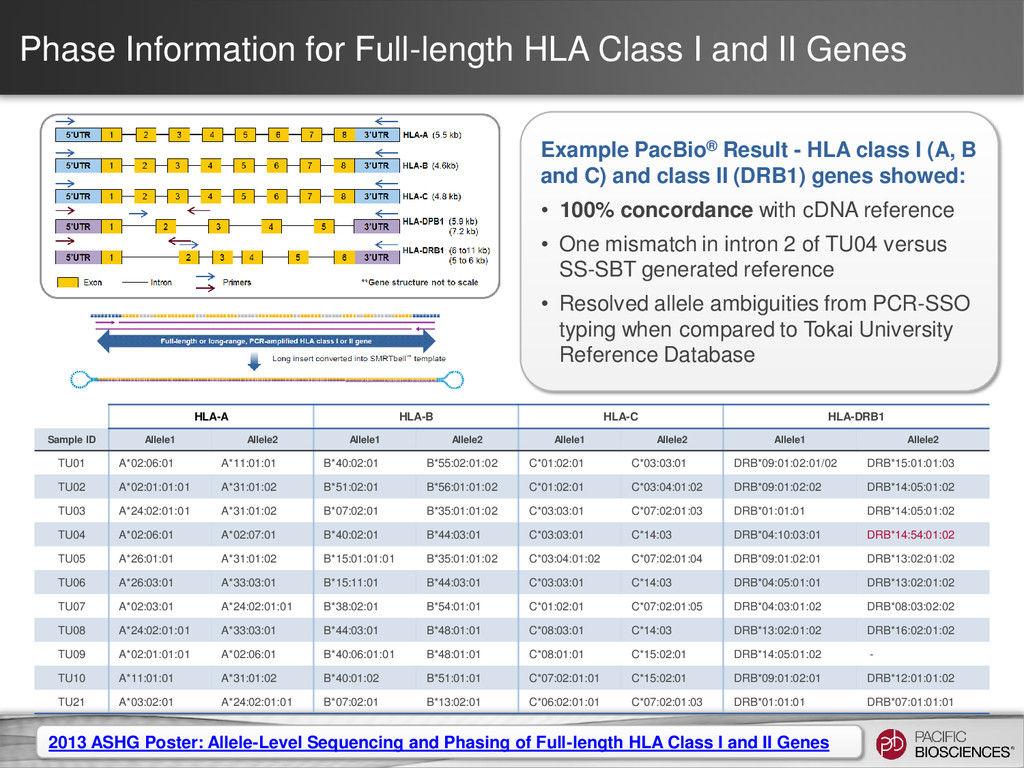
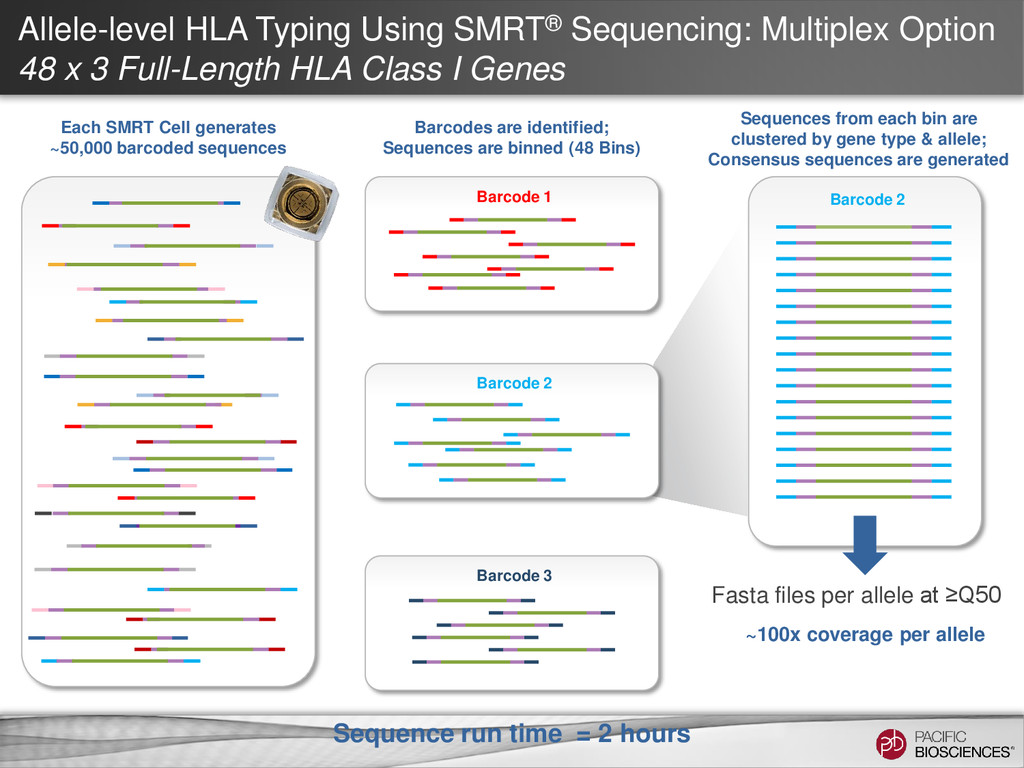
3 Full-Length HLA Class I Genes Each SMRT Cell generates ~50,000 barcoded sequences Barcode 2 Sequences from each bin are clustered by gene type & allele; Consensus sequences are generated ~100x coverage per allele Fasta files per allele at ≥Q50 Barcode 3 Barcode 1 Barcodes are identified; Sequences are binned (48 Bins) Barcode 2 Sequence run time = 2 hours
~50,000 Barcoded Sequences Barcode 2 Sequences from Each Bin Are Aligned and Bases Are Called ~100x coverage per clone • Q50 accuracy at ~30x coverage Single Fasta file at ≥Q50 Barcode 3 Barcode 1 Barcodes Are Identified and Sequences Are Binned (384 Bins) Barcode 2
of data at differing levels of coverage • At 45X coverage, errors detected with a frequency of 10-5 • Above 50X coverage, no errors detected in ~700 kb of sequence 6.4 X 10-5 2.1 X 10-5 Error Rates
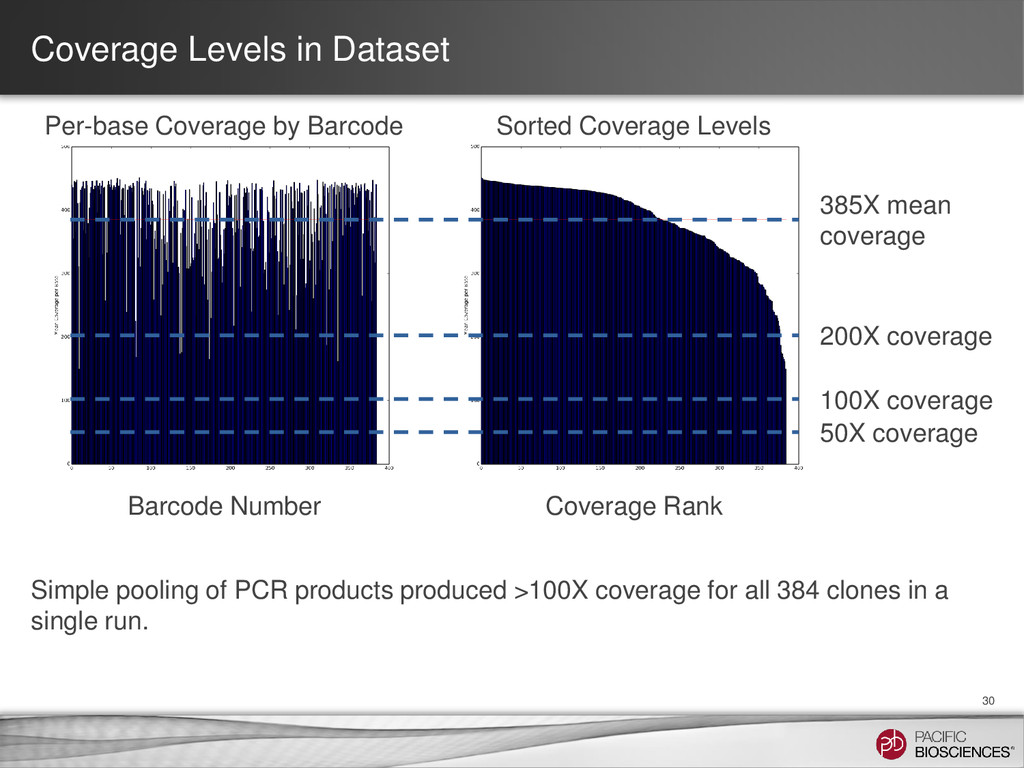
mean coverage Per-base Coverage by Barcode Barcode Number Coverage Rank Sorted Coverage Levels Simple pooling of PCR products produced >100X coverage for all 384 clones in a single run. 50X coverage
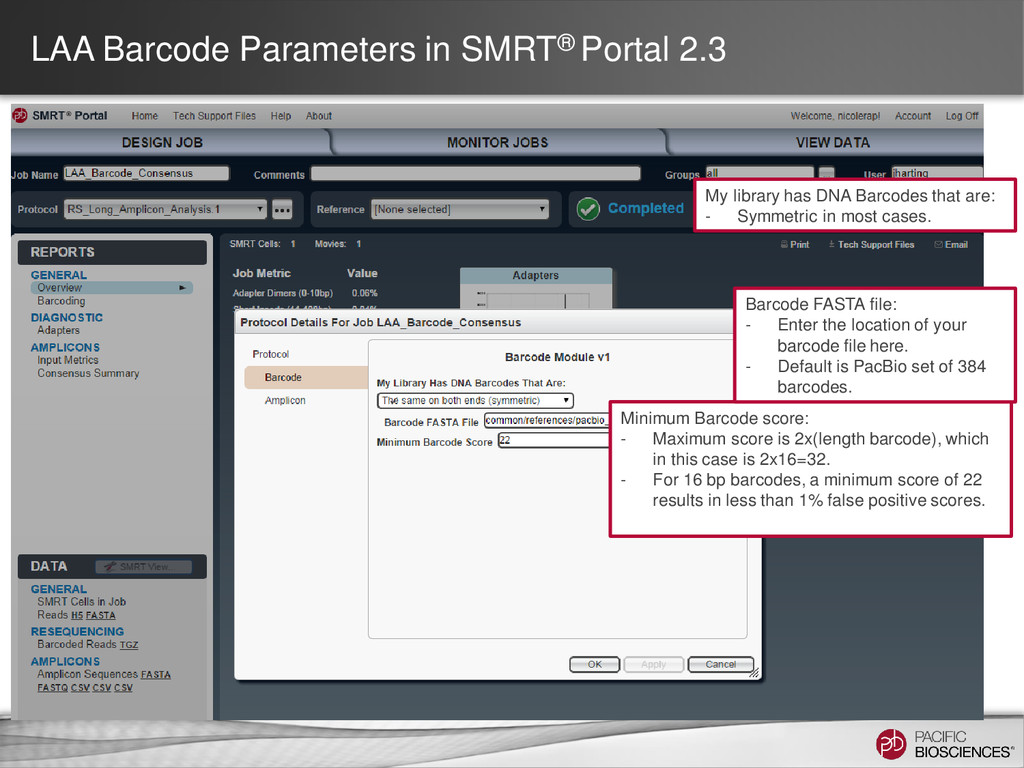
DNA Barcodes that are: - Symmetric in most cases. Minimum Barcode score: - Maximum score is 2x(length barcode), which in this case is 2x16=32. - For 16 bp barcodes, a minimum score of 22 results in less than 1% false positive scores. Barcode FASTA file: - Enter the location of your barcode file here. - Default is PacBio set of 384 barcodes.
- Set to 80% of your insert size Coarse Cluster Subreads by Gene Family: - Keep clicked for HLA - Unclick for amplicon consensus calling. Maximum number of subreads: - Set to 200 Phase Alleles: - Keep clicked for HLA or other applications where you expect 2 alleles. - Unclick for amplicon with a single allele.
Pacific Biosciences, the Pacific Biosciences logo, PacBio, SMRT, SMRTbell, and Iso-Seq are trademarks of Pacific Biosciences in the United States and/or other countries. All other trademarks are the sole property of their respective owners.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}