Matthew W. Hahn1,2,3 and Luay Nakhleh4,5 1Department of Biology, Indiana University, Bloomington, Indiana 47405 2School of Informatics and Computing, Indiana University, Bloomington, Indiana 47405 3E-mail:
[email protected] 4Department of Computer Science, Rice University, Houston, Texas 77005 5BioSciences, Rice University, Houston, Texas 77005 Received November 15, 2015 Accepted November 30, 2015 Phylogenomics has largely succeeded in its aim of accurately inferring species trees, even when there are high levels of discordance among individual gene trees. These resolved species trees can be used to ask many questions about trait evolution, including the direction of change and number of times traits have evolved. However, the mapping of traits onto trees generally uses only a single representation of the species tree, ignoring variation in the gene trees used to construct it. Recognizing that genes underlie traits, these results imply that many traits follow topologies that are discordant with the species topology. As a consequence, standard methods for character mapping will incorrectly infer the number of times a trait has evolved. This phenomenon, dubbed “hemiplasy,” poses many problems in analyses of character evolution. Here we outline these problems, explaining where and when they are likely to occur. We offer several ways in which the possible presence of hemiplasy can be diagnosed, and discuss multiple approaches to dealing with the problems presented by underlying gene tree discordance when carrying out character mapping. Finally, we discuss the implications of hemiplasy for general phylogenetic inference, including the possible drawbacks of the widespread push for “resolved” species trees. Points of View Syst. Biol. 67(6):1091–1109, 2018 © The Author(s) 2018. Published by Oxford University Press, on behalf of the Society of Systematic Biologists. All rights reserved. For Permissions, please email:
[email protected] DOI:10.1093/sysbio/syy031 Advance Access publication April 25, 2018 Rethinking phylogenetic comparative methods JOSEF C. UYEDA 1,∗, ROSANA ZENIL-FERGUSON 2,3, AND MATTHEW W. PENNELL 4 1Department of Biological Sciences, Virginia Polytechnic Institute and State University, 926 West Campus Drive, Blacksburg, 2Department of Biological Sciences, University of Idaho, 875 Perimeter Drive, Moscow, ID 83844 USA; 3Department of Ecology, Evolution and Behavior, University of Minnesota, 1479 Gortner Avenue, St. Paul, MN 55108 4Department of Zoology and Biodiversity Research Centre, University of British Columbia, #4200-6700 University Blvd., Vancouver ∗Correspondence to be sent to: Department of Biological Sciences, Virginia Polytechnic Institute and State University, Blacksbur E-mail:
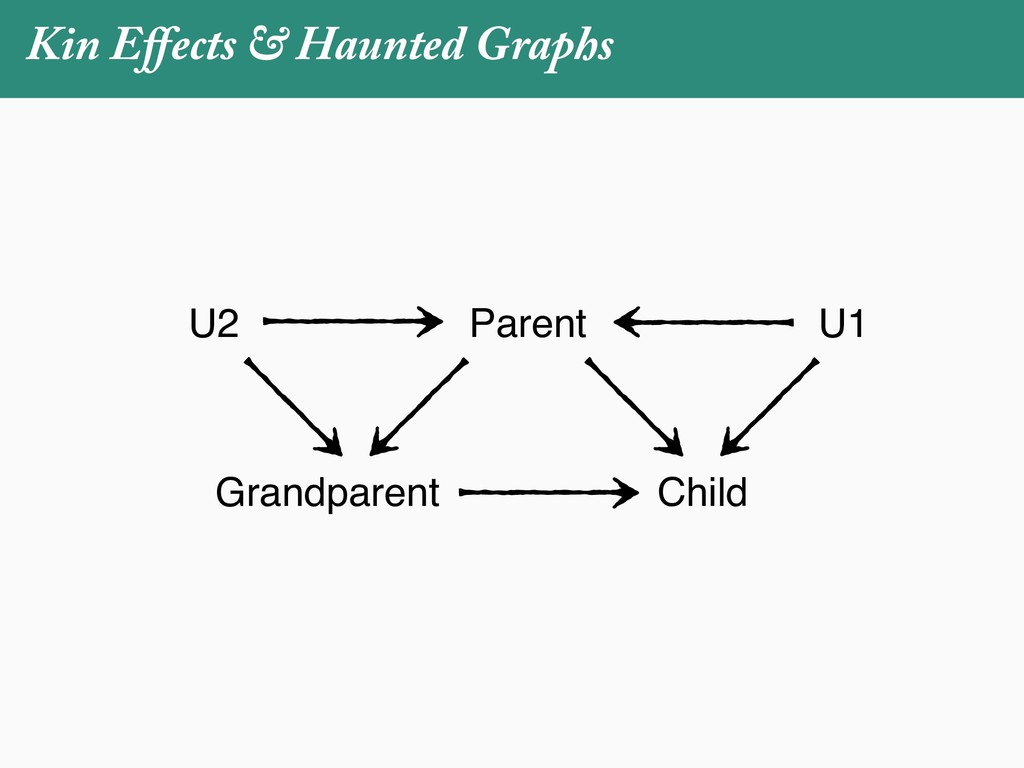
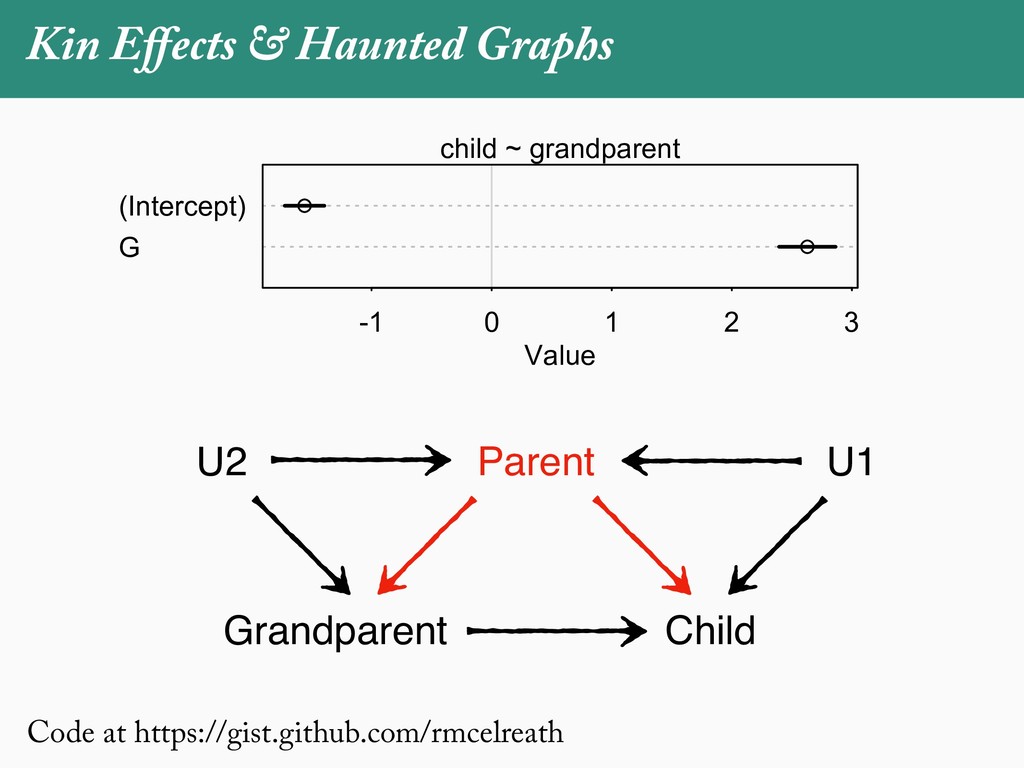
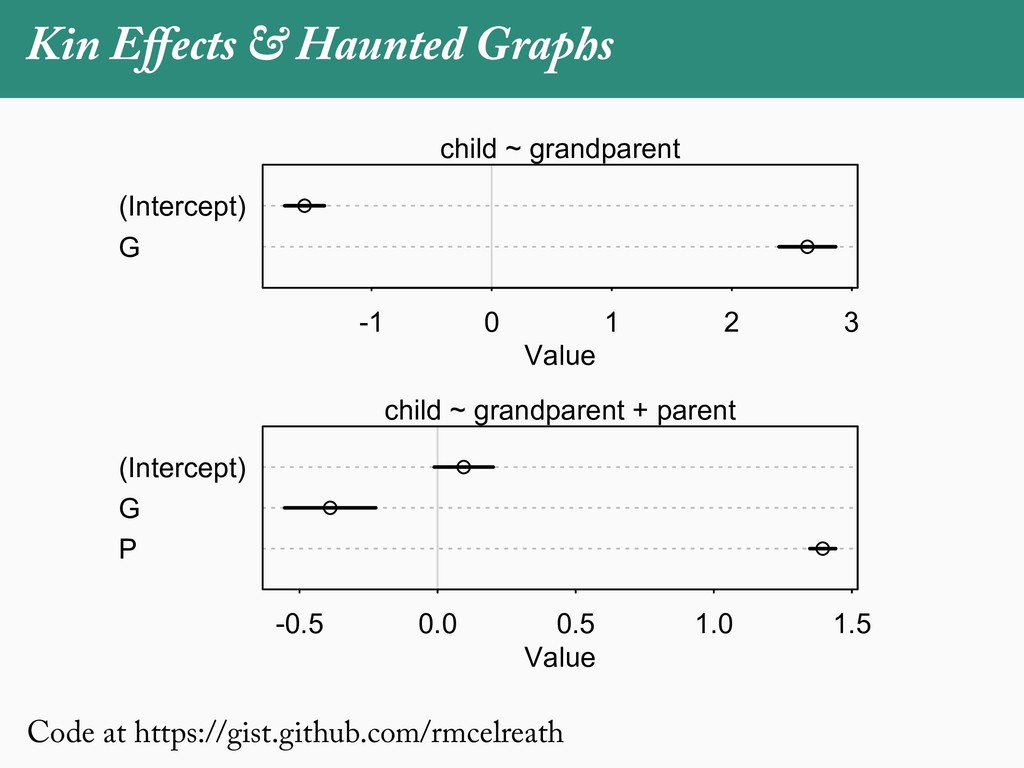
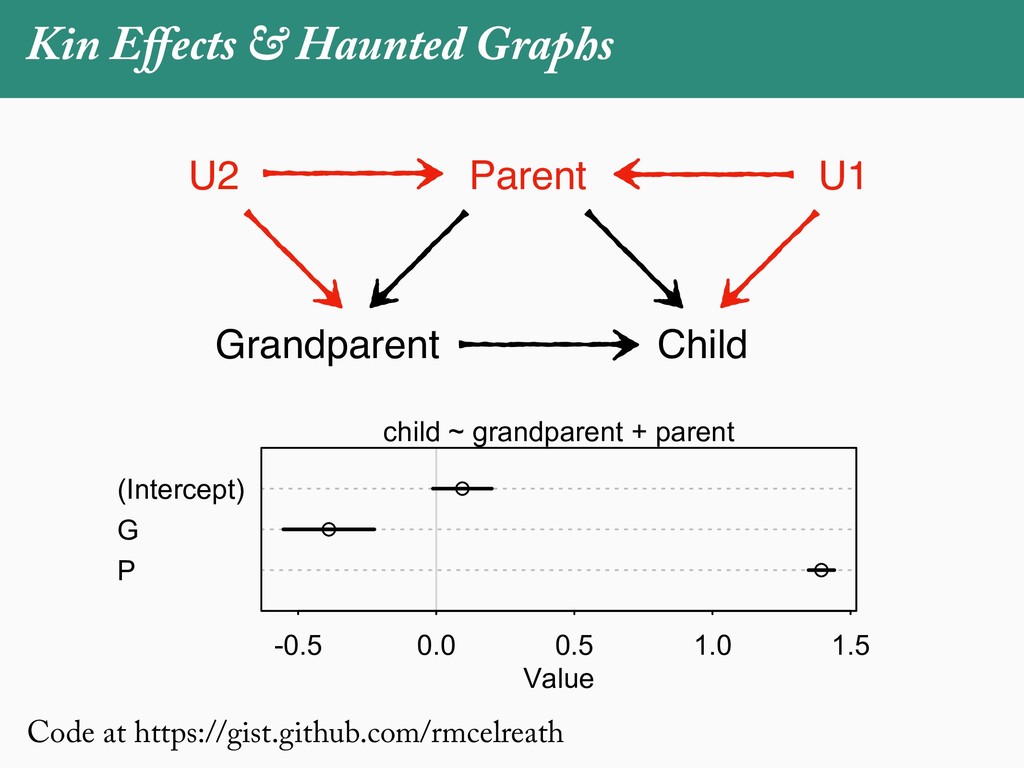
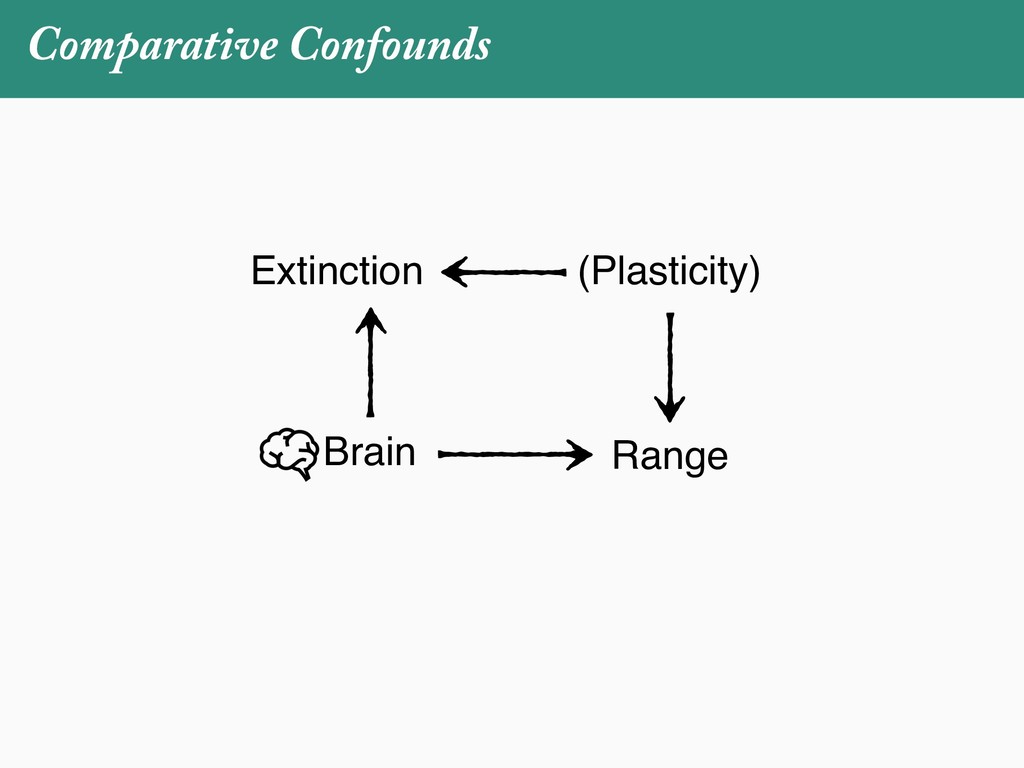
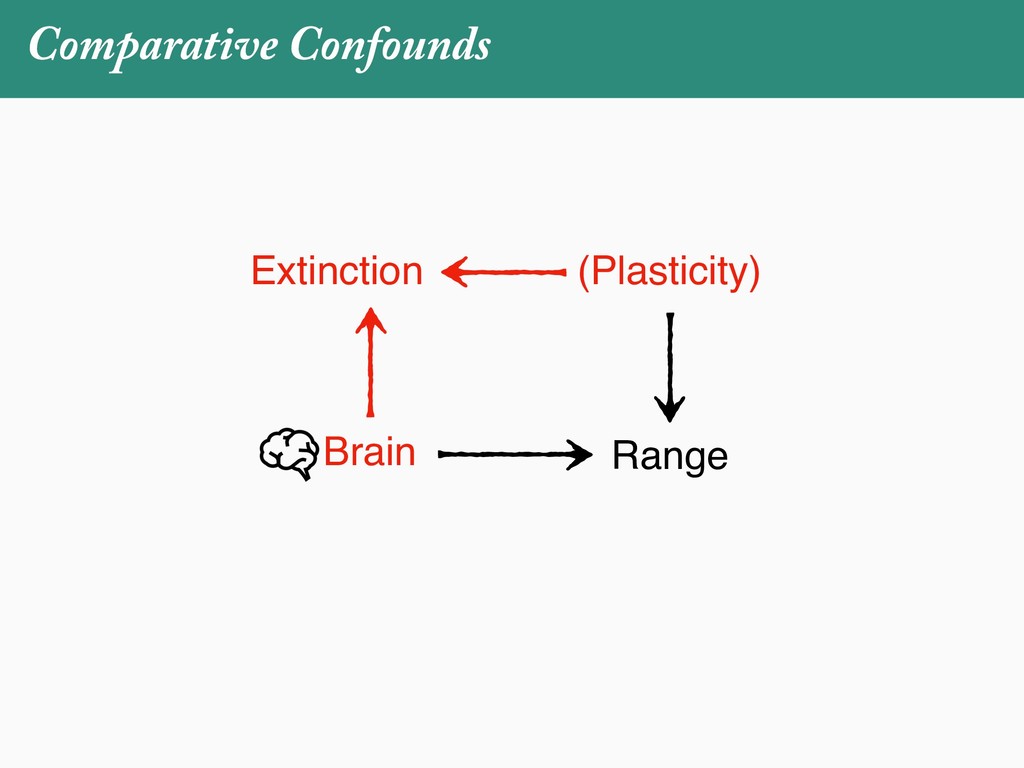
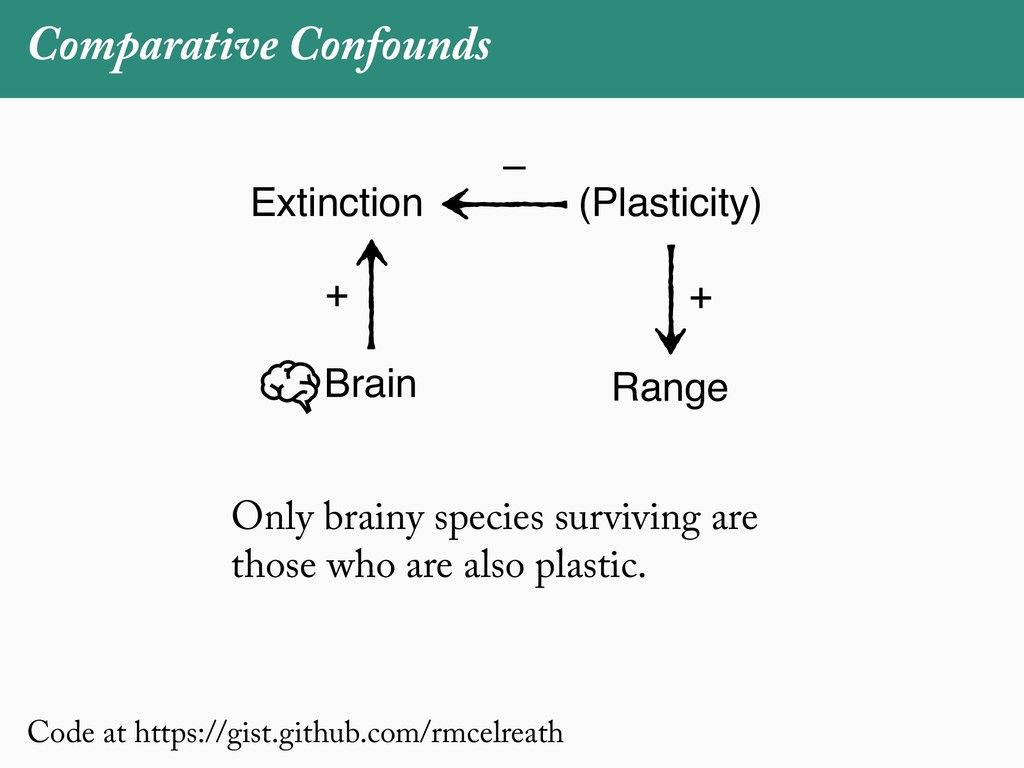
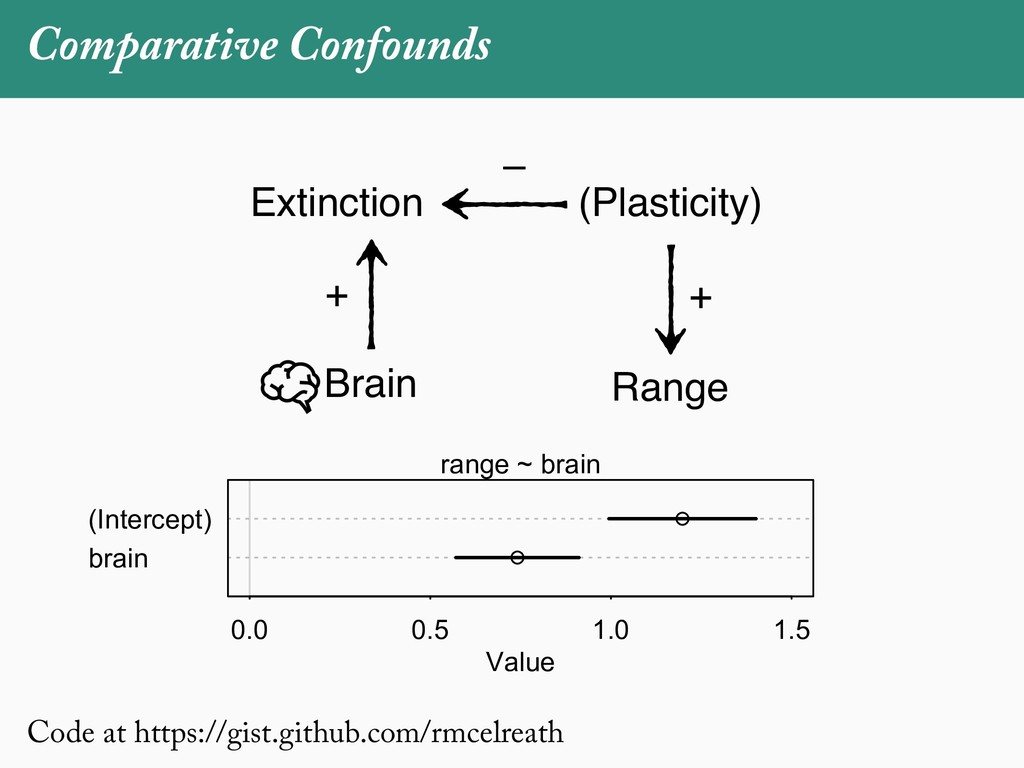
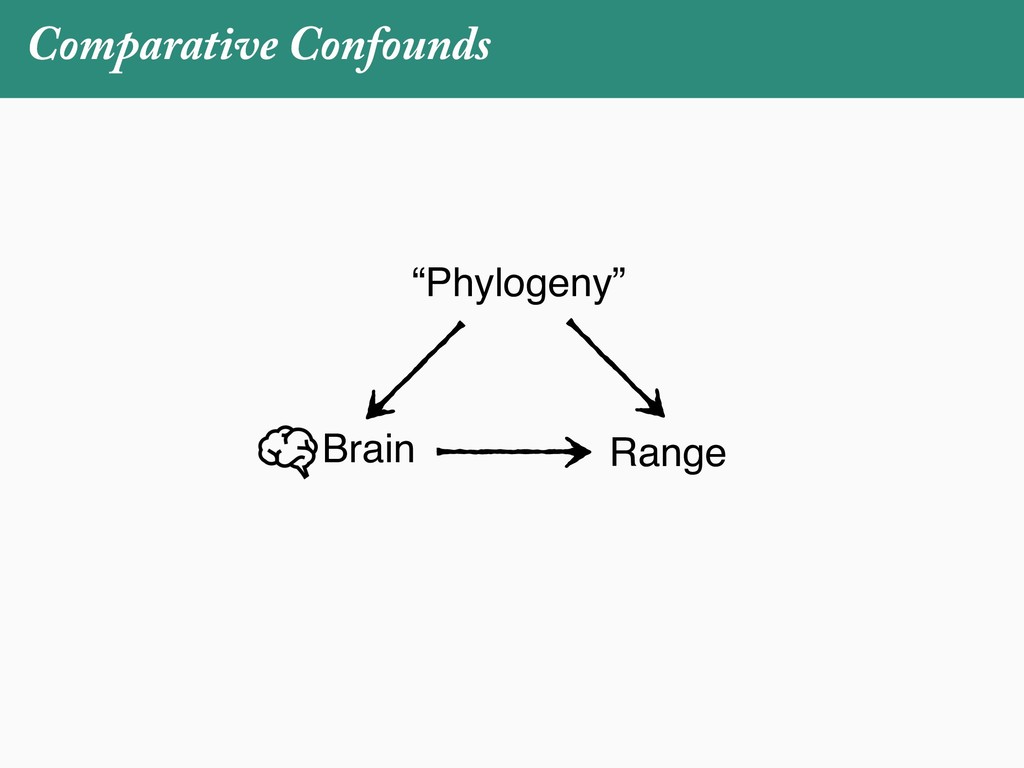
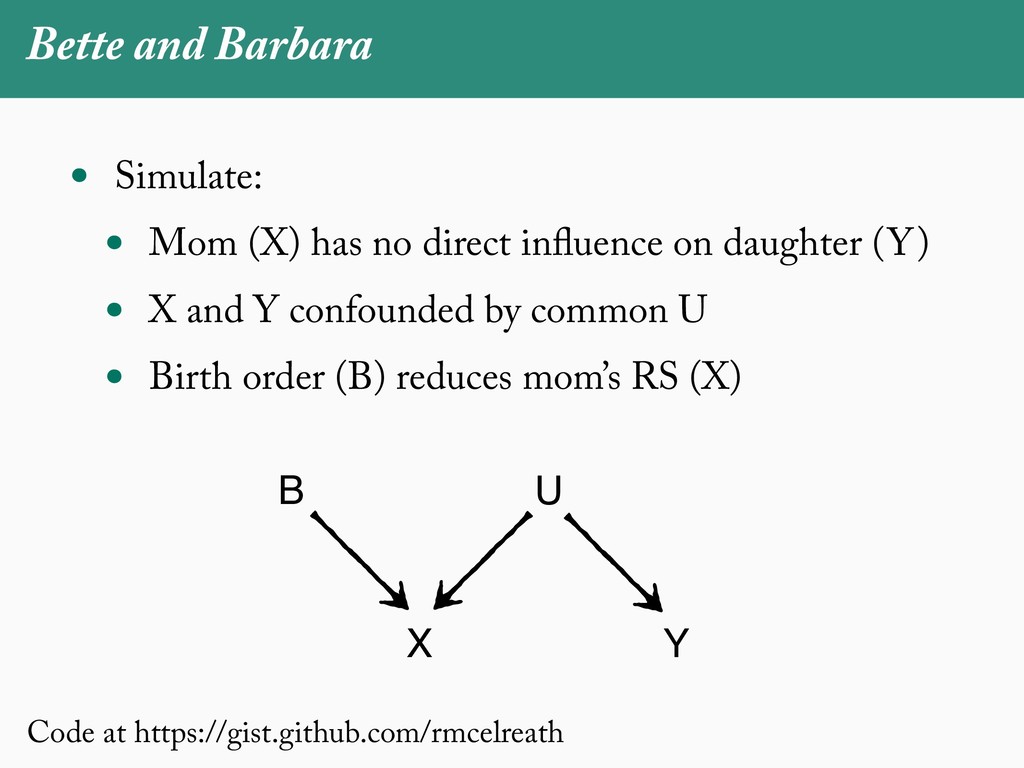
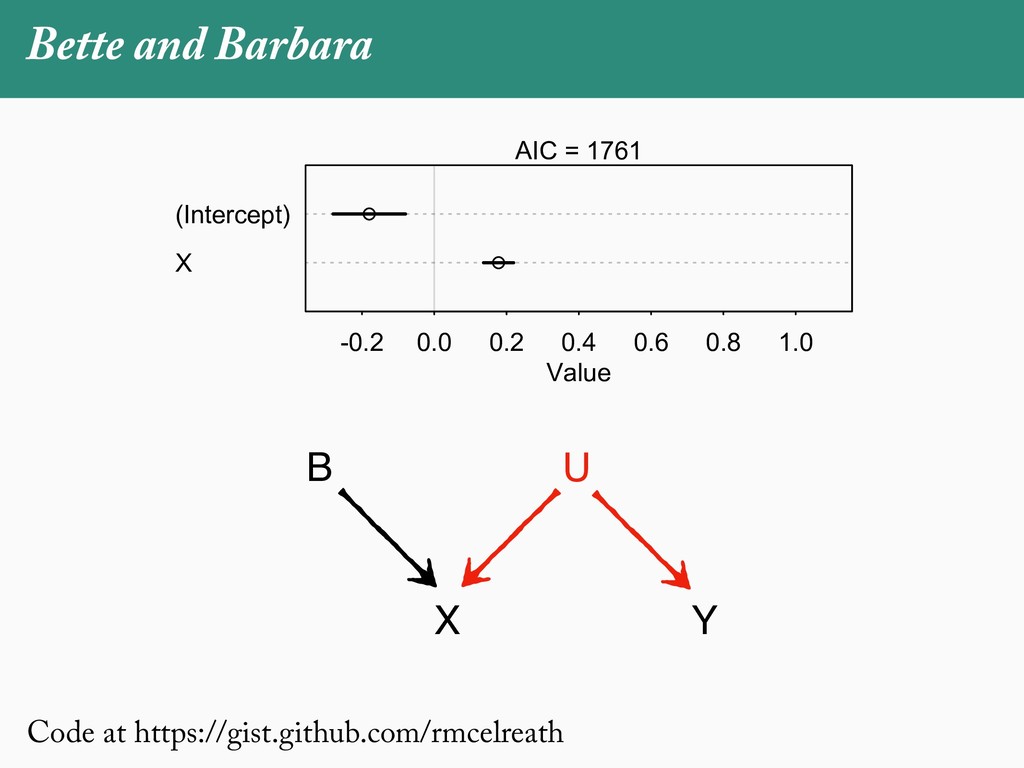
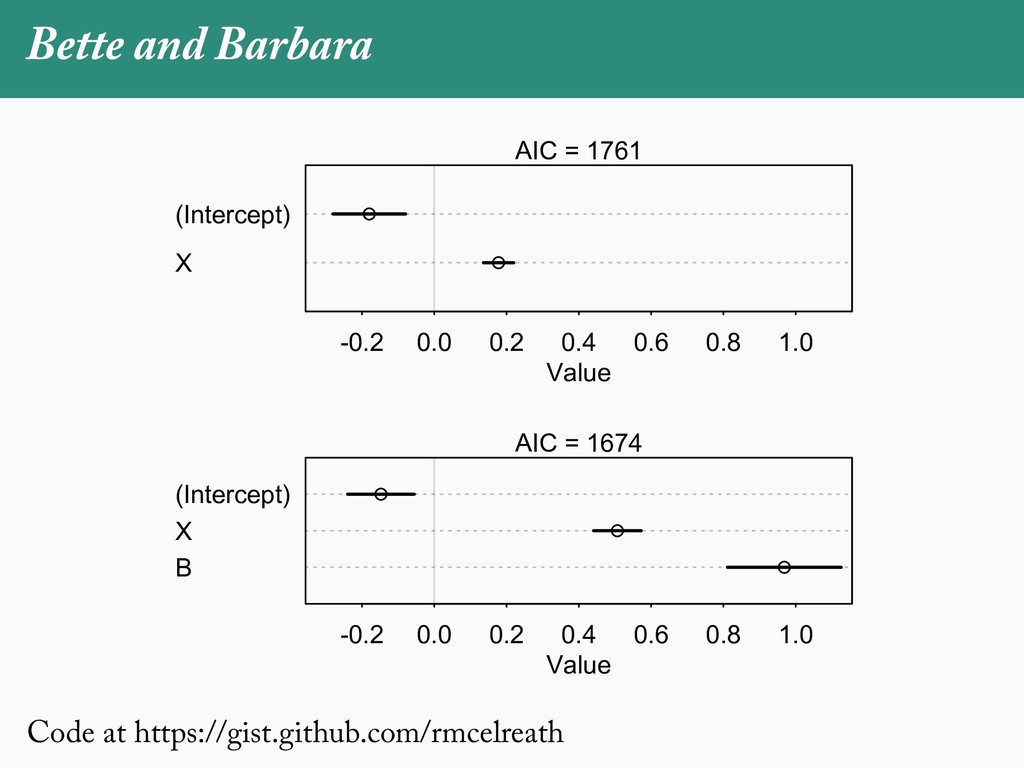
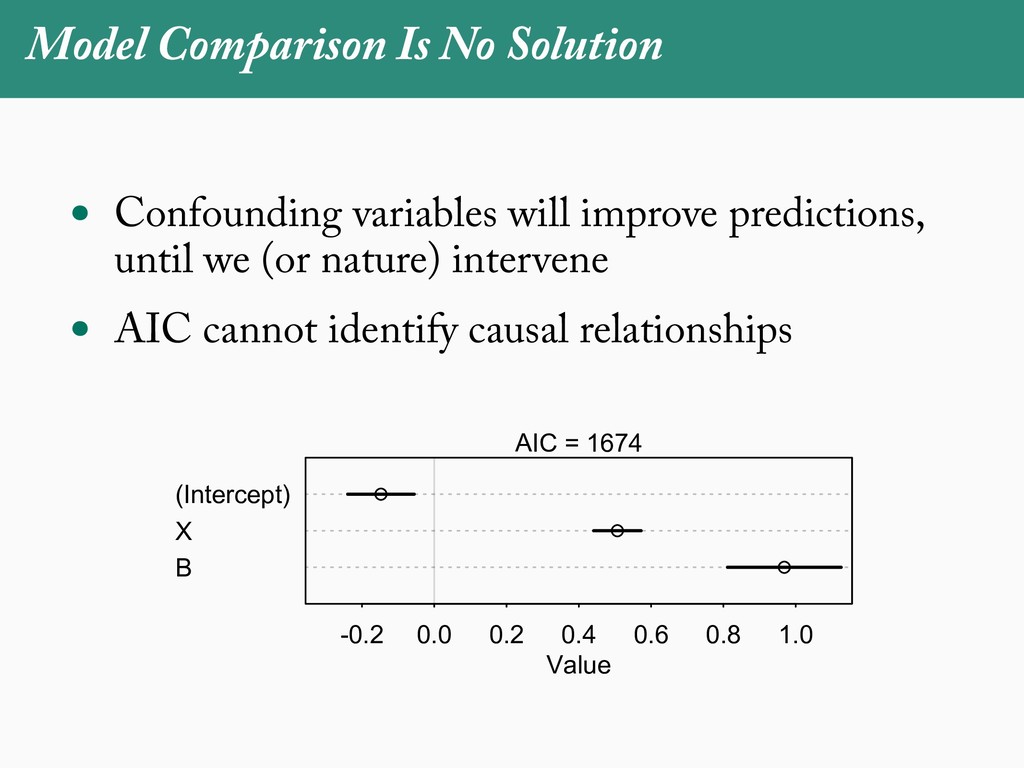
[email protected]. Received: 21 November 2017; reviews returned: 17 March 2018; accepted: 17 April 2018 Associate Editor: Nicholas Matzke Abstract.—As a result of the process of descent with modification, closely related species tend to be similar to one a myriad different ways. In statistical terms, this means that traits measured on one species will not be independ measured on others. Since their introduction in the 1980s, phylogenetic comparative methods (PCMs) have been a solution to this problem. In this article, we argue that this way of thinking about PCMs is deeply misleading. N this sowed widespread confusion in the literature about what PCMs are doing but has led us to develop metho susceptible to the very thing we sought to build defenses against—unreplicated evolutionary events. Through Studies, we demonstrate that the susceptibility to singular events is indeed a recurring problem in comparative b ited by: YS MANUSCRIPT CATEGORY: Points of View 04 SYSTEMATIC BIOLOGY VOL. 67 A B
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}