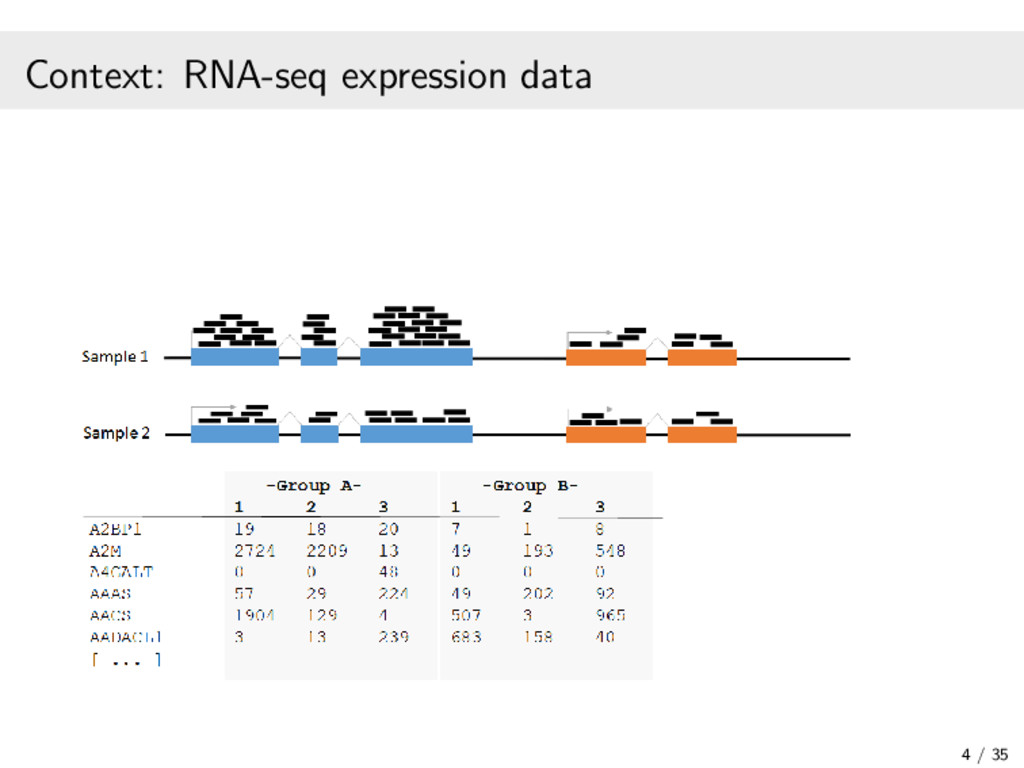
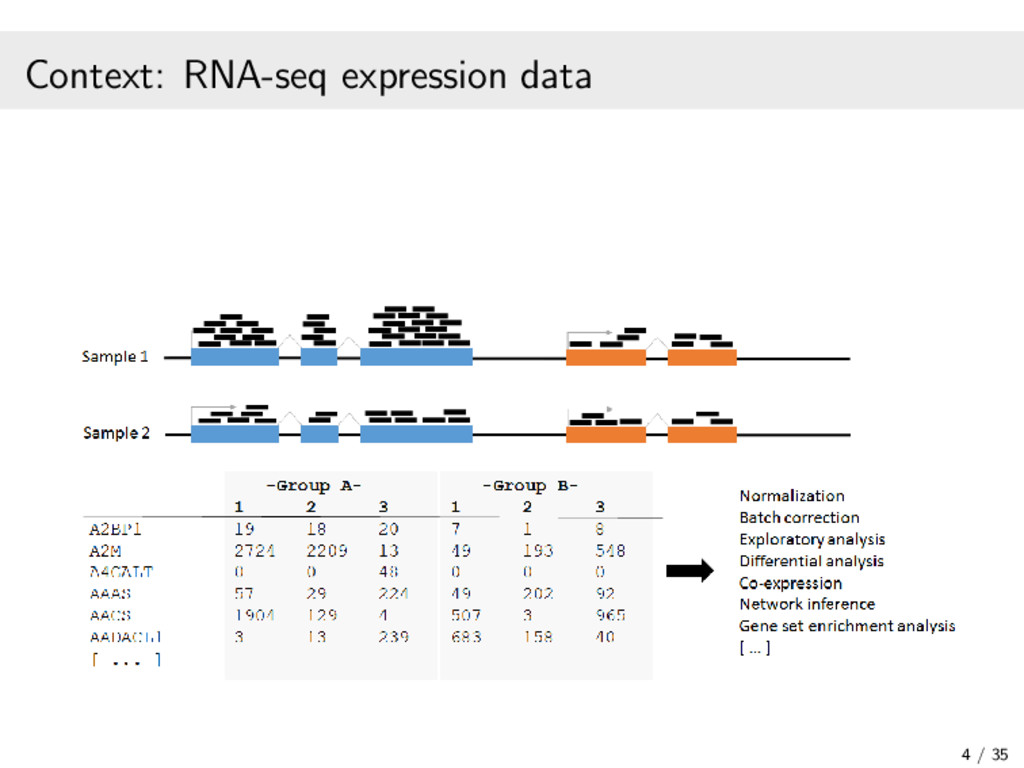
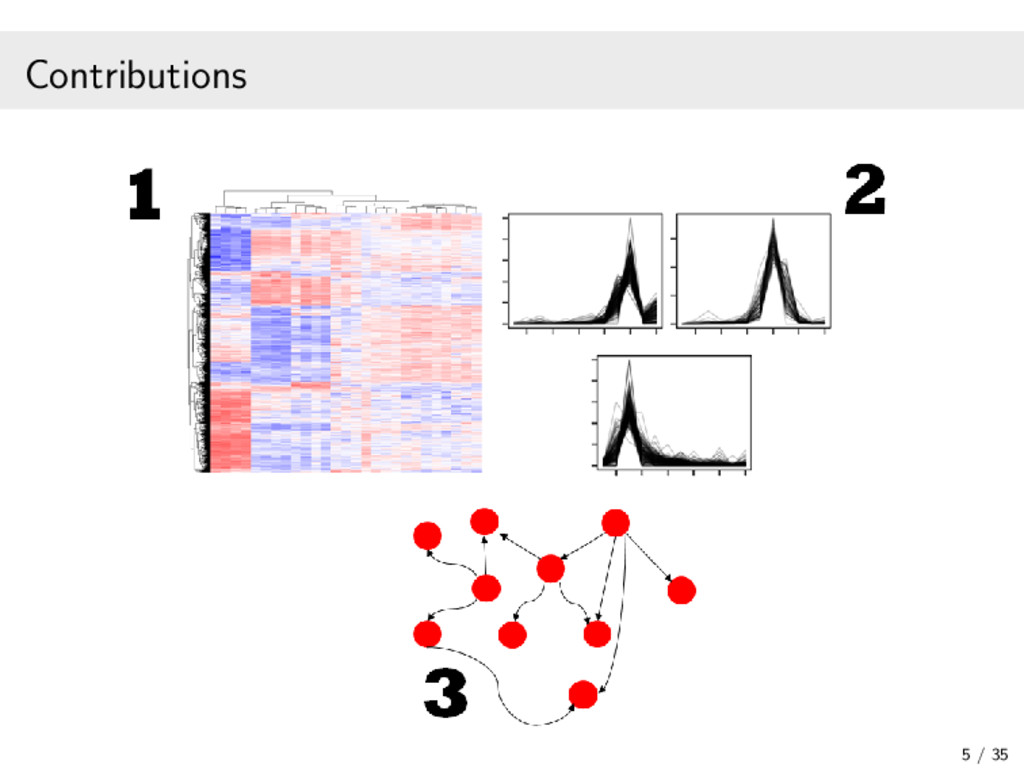
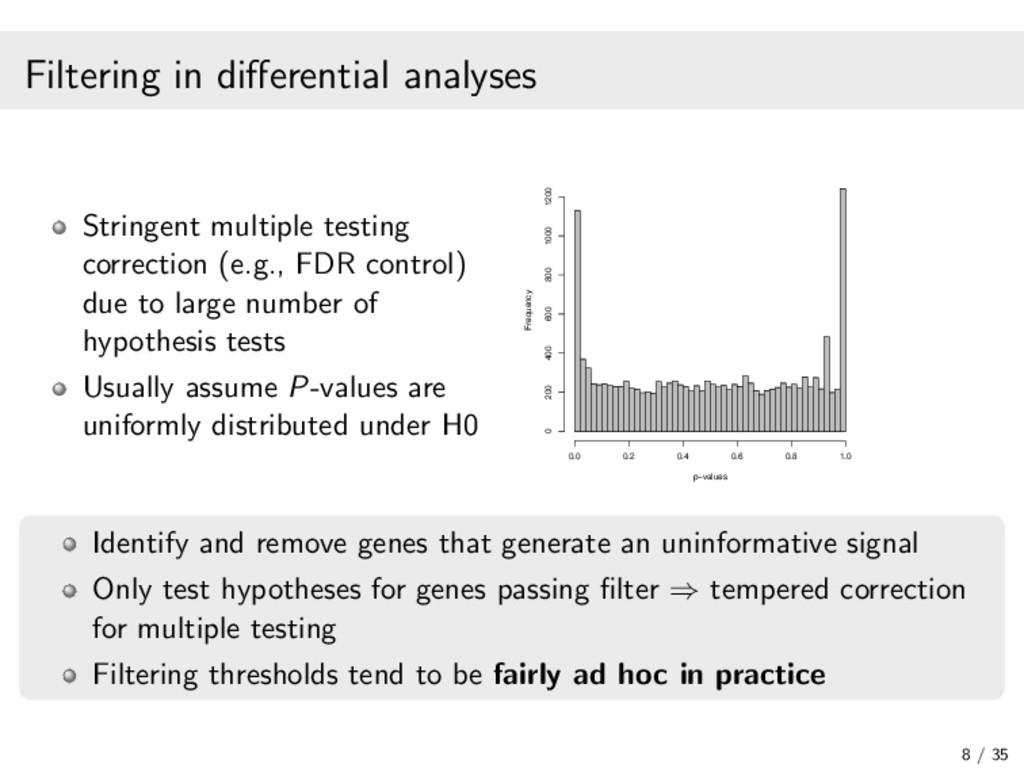
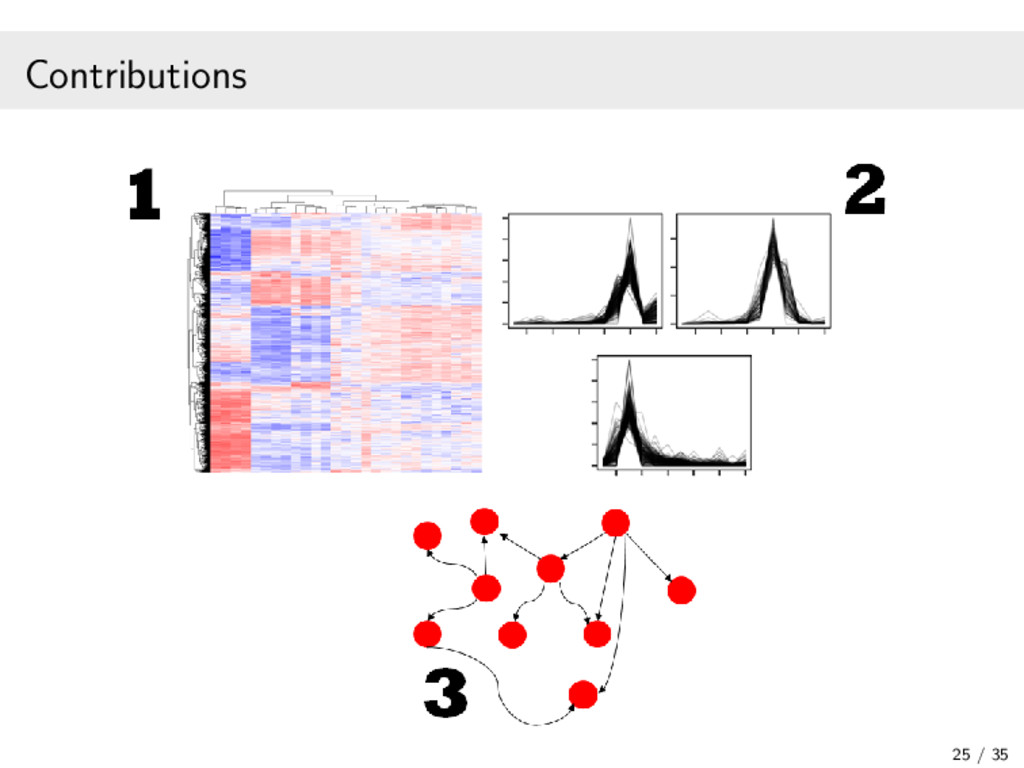
In recent years, high-throughput sequencing (HTS) has become an essential tool for genomic and transcriptomic studies. In particular, the use of HTS to directly sequence reverse-transcribed RNA molecules, known as RNA sequencing (RNA-seq), has revolutionized the study of gene expression. In turn, a great deal of methodological research has focused on developing analysis pipelines that are well suited to the characteristics of RNA-seq data. In this work, I focus on methodological contributions to three analytical goals: (1) the powerful detection of differentially expressed genes from RNA-seq data, in particular through a data-based filter for weakly expressed genes and a P-value combination approach for data arising from multiple related studies; (2) the identification of clusters of co-expressed genes from RNA-seq data using finite mixture models, appropriately chosen transformations, and adapted model selection criteria; and (3) the inference of gene regulatory networks from observational RNA-seq data or arbitrarily complex gene knockout expression data. In addition, I will present some of the open-source software packages I have developed and continue to maintain for the implementation of our proposed statistical methods. Finally, I will discuss some related research perspectives regarding methodological developments for multi-omics data integration.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}