Presentation at MiMo workshop on mixture models (April 8, 2021)
https://mimo2021.sciencesconf.org
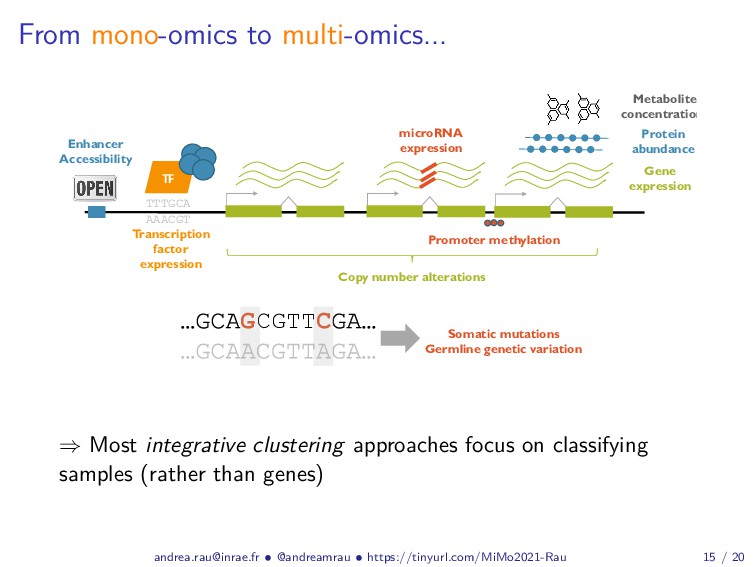
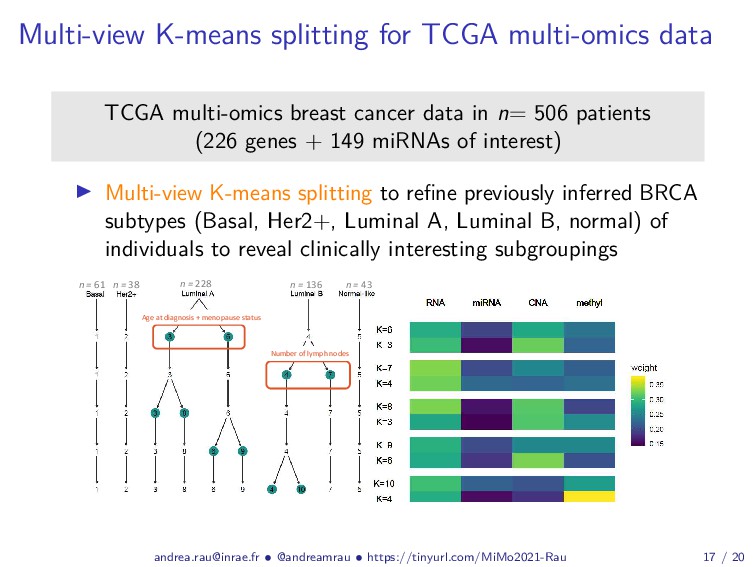
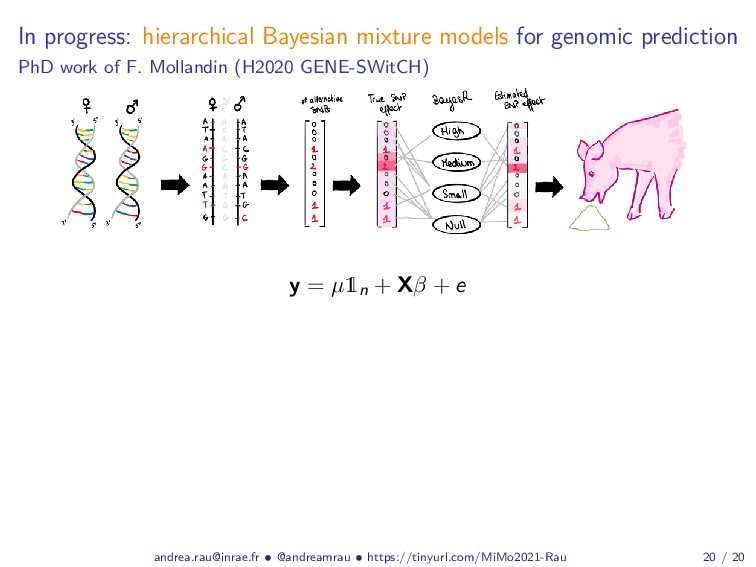
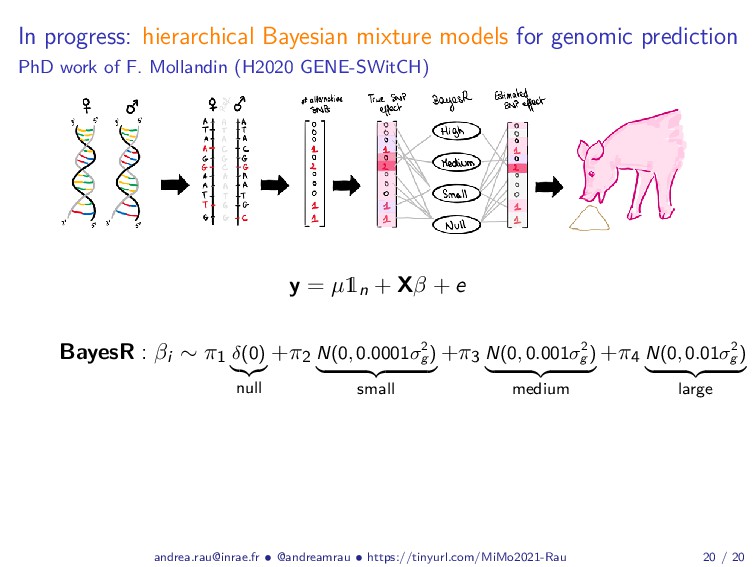
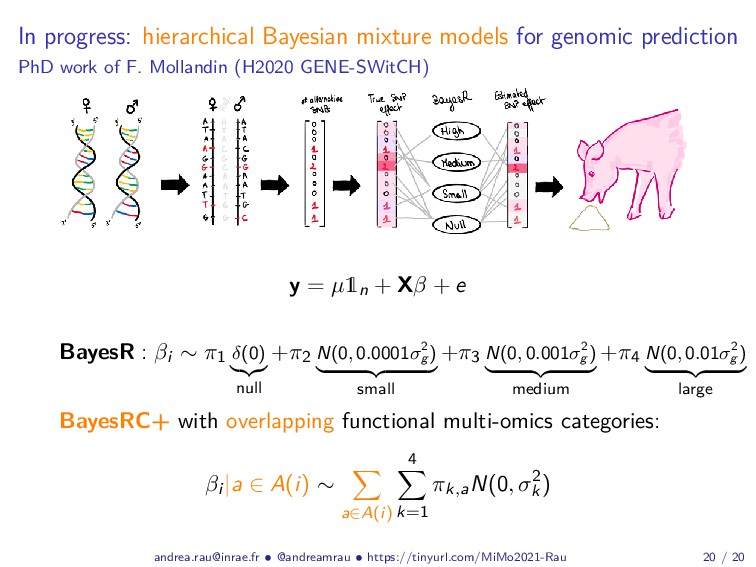
Abstract: Complex studies of transcriptome dynamics are now routinely carried out using RNA sequencing (RNA-seq). A common goal in such studies is to identify groups of co-expressed genes that share similar expression profiles across several treatment conditions, time points, or tissues. These co-expression analyses serve both as an exploratory visualization tool as well as a hypothesis-generating tool for poorly annotated genes. In this talk, I will discuss some of the mixture-model based approaches we have developed in recent years for RNA-seq co-expression analysis, with a particular focus on the practical issues surrounding the use of such approaches and their implementation within the R/Bioconductor package ecosystem. Finally, as studies with matched multi-view data (i.e., at different biological levels of molecular information) are becoming increasingly common, I will also briefly discuss our recent work to integratively use multiple data views to aggregate or split existing clusters from multi-omics data.
{kind=link}
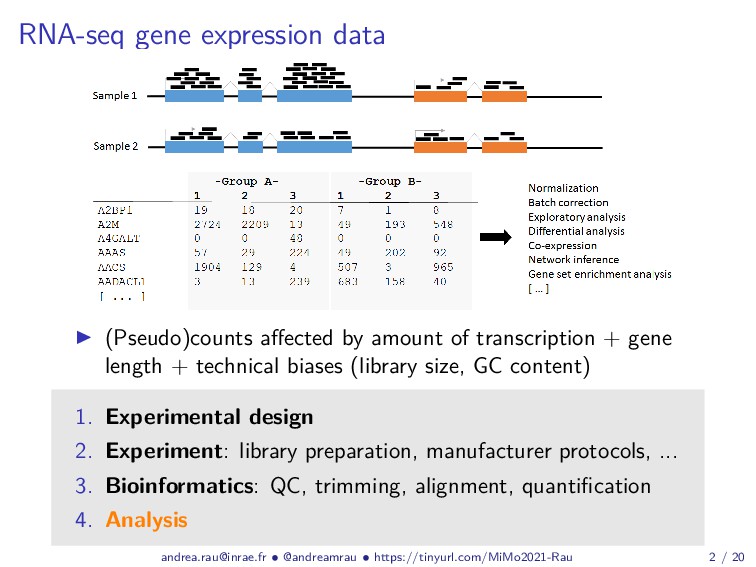{kind=link}
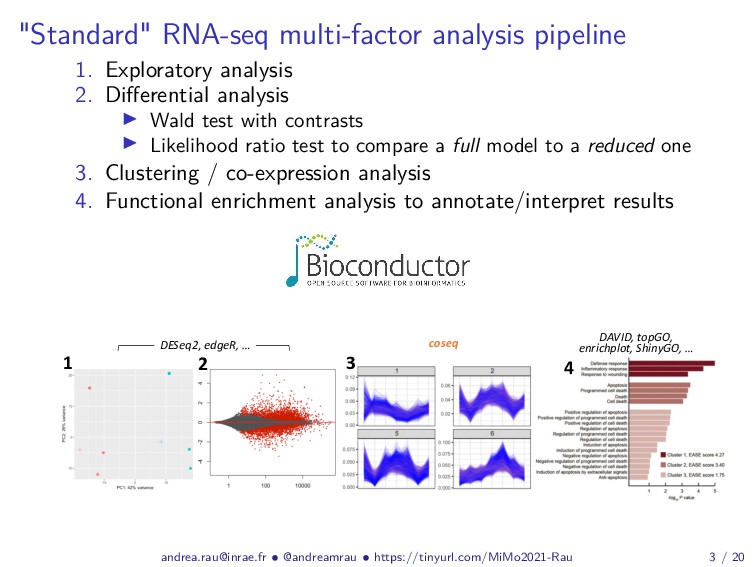{kind=link}
{kind=link}
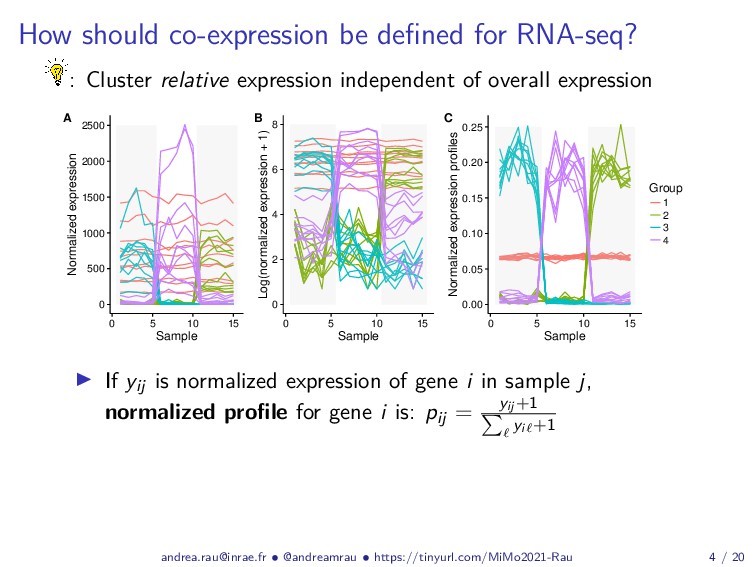{kind=link}
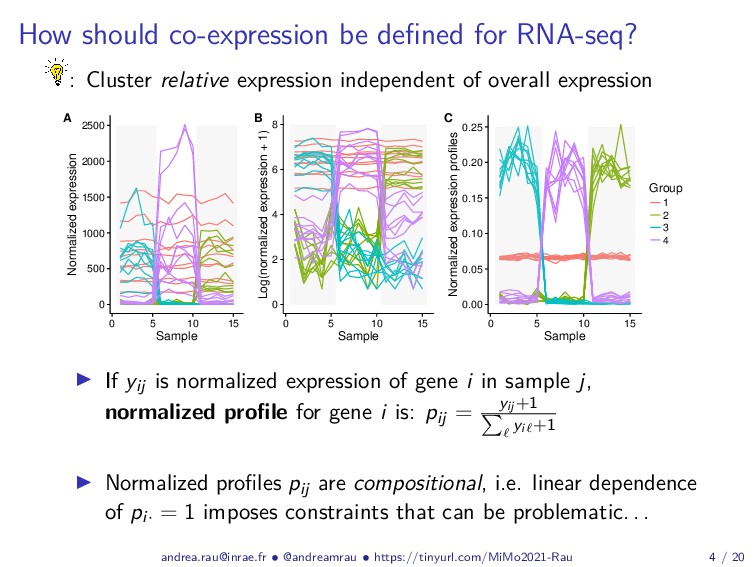{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Some thoughts about software usability [email protected] • @andreamrau • https://tinyurl.com/MiMo2021-Rau](https://files.speakerdeck.com/presentations/5204058366664477a86cc0b5109e856f/slide_21.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}