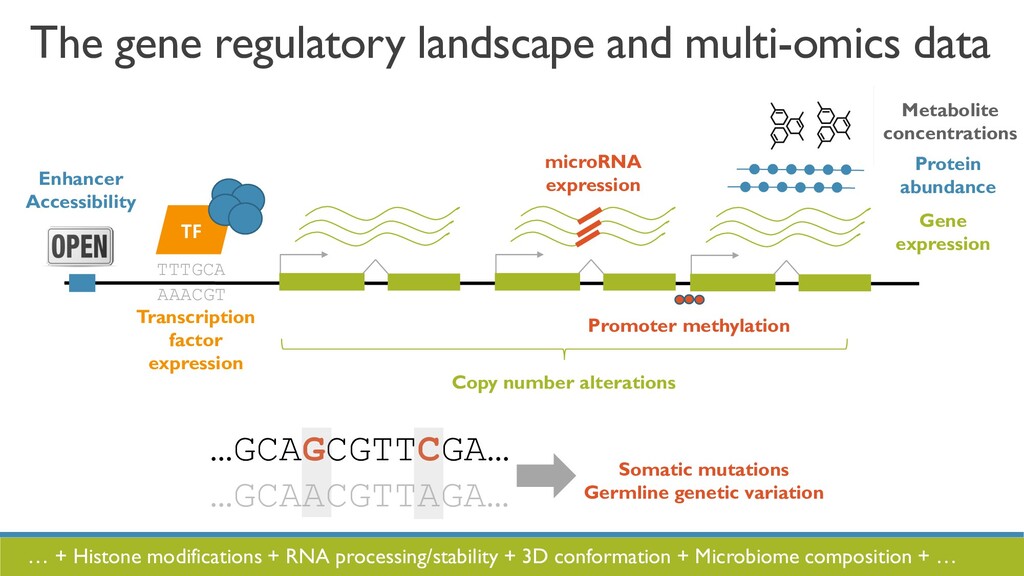
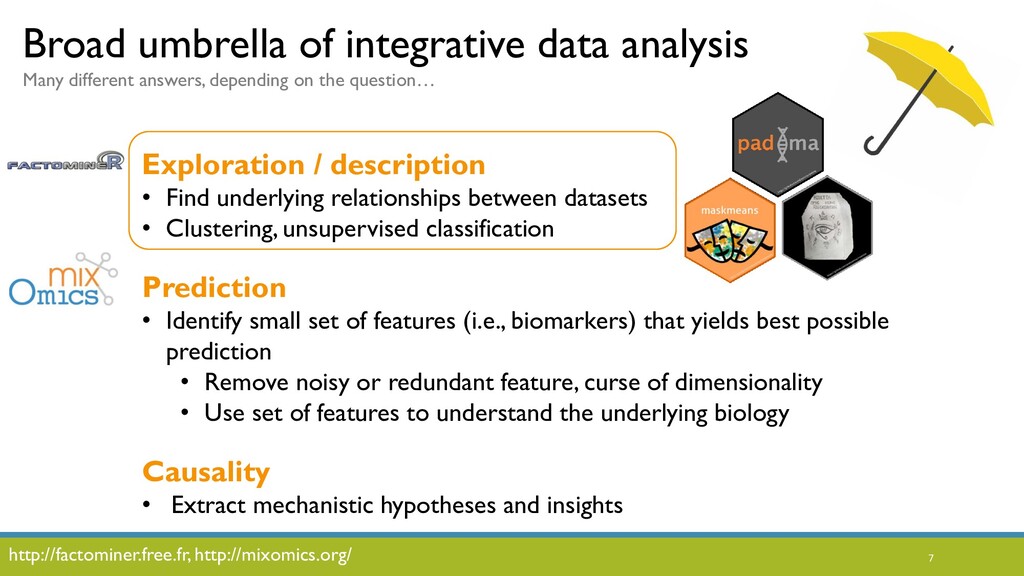
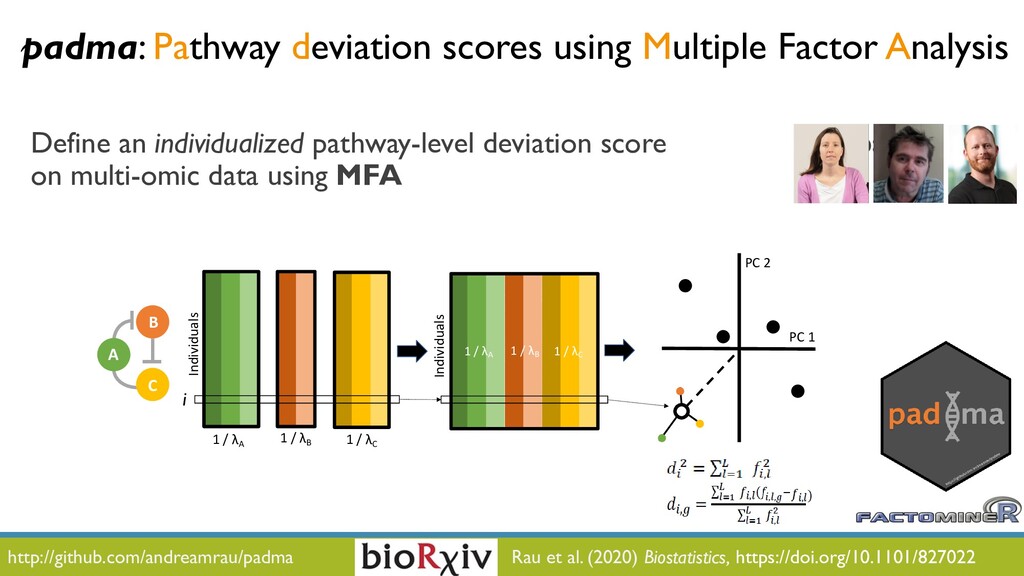
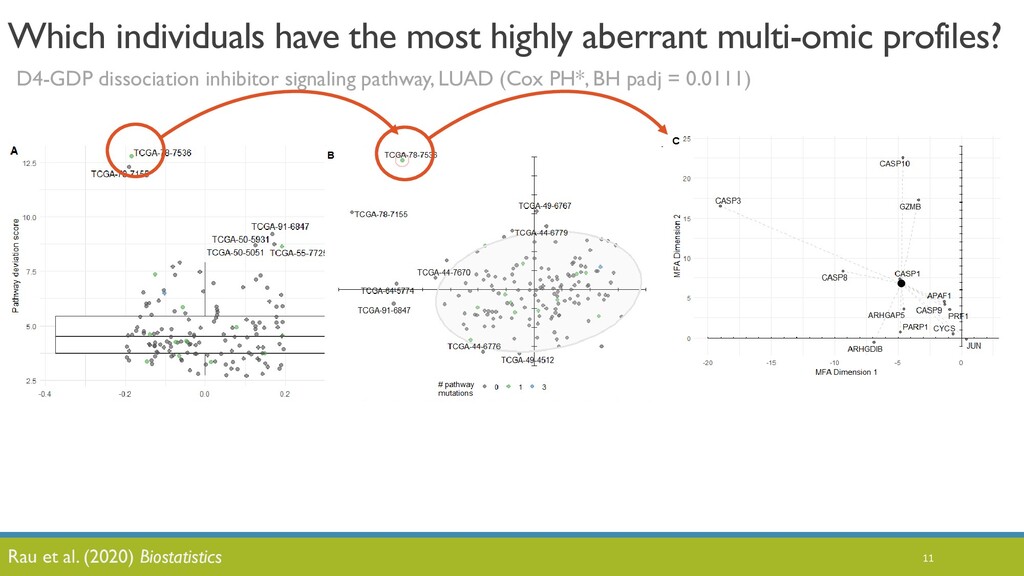
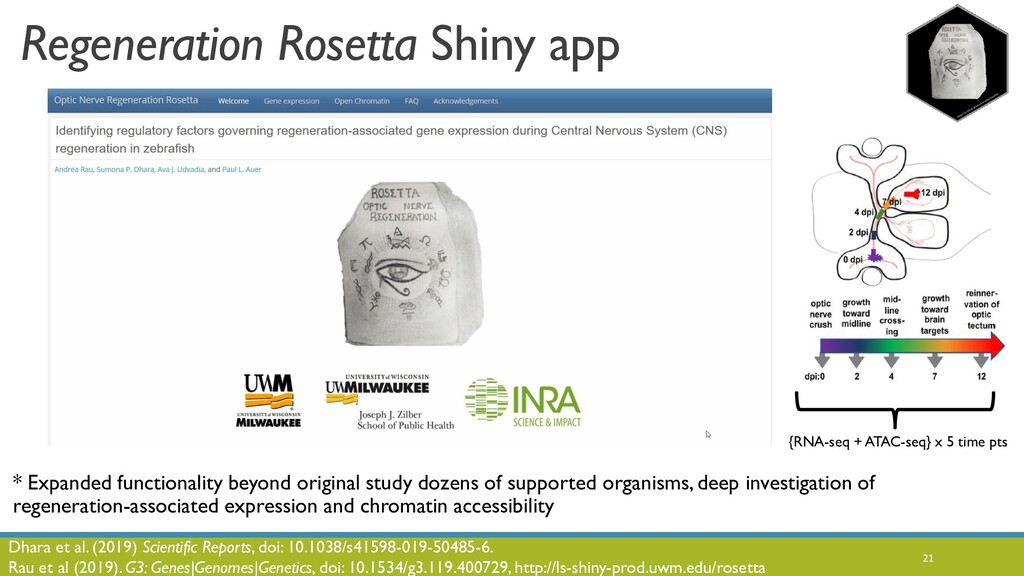
JOBIM 2020 keynote talk: The increased availability and affordability of high-throughput sequencing technologies in recent years has facilitated the use of multi-omic studies to expand and enrich our understanding of complex biological systems. However, defining a holistic and meaningful way to exploit these heterogeneous and multi-faceted ‘omics data can be complicated by several major obstacles. These include the unknown hierarchy and potentially ambiguous relationships among different sources of data, the explosion in data dimension, issues due to batch effects and quality control, potentially incomplete or missing data, limited sample sizes, and the occasional difficulty in posing well-defined and answerable research questions of such data. In light of these challenges, in this talk I will provide an overview of some of our methodological contributions to integrative multi-omic analyses, and I will discuss how the development of interactive tools can be a useful addition to the multi-omic analysis toolbox.
https://jobim2020.sciencesconf.org/
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}