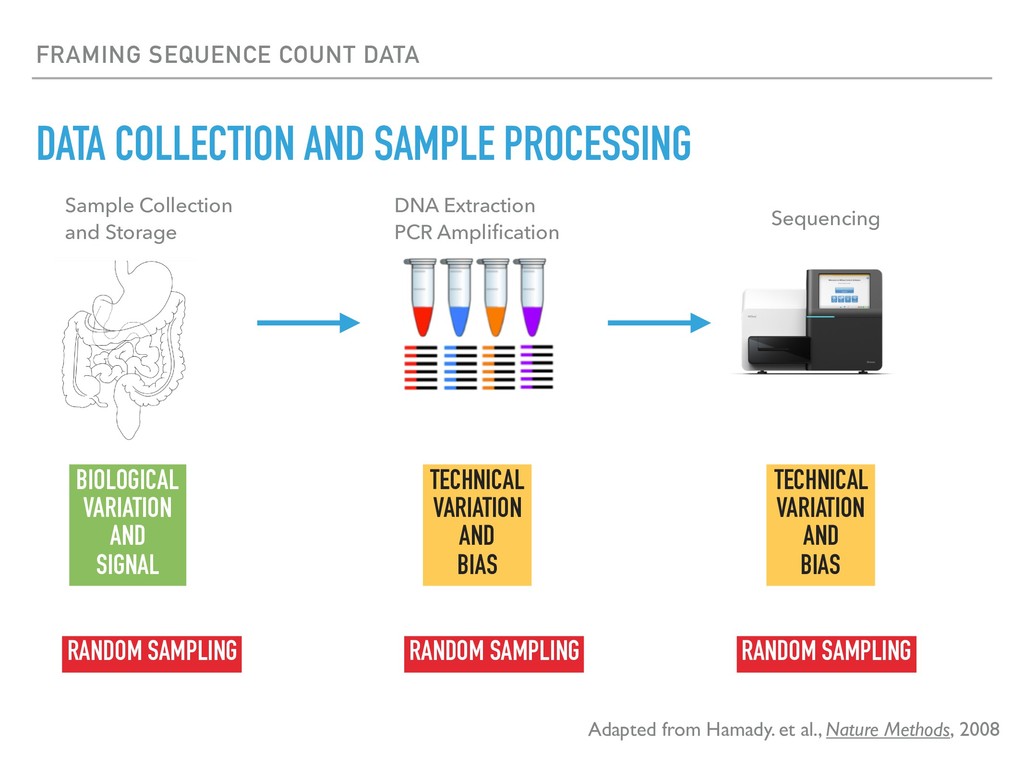
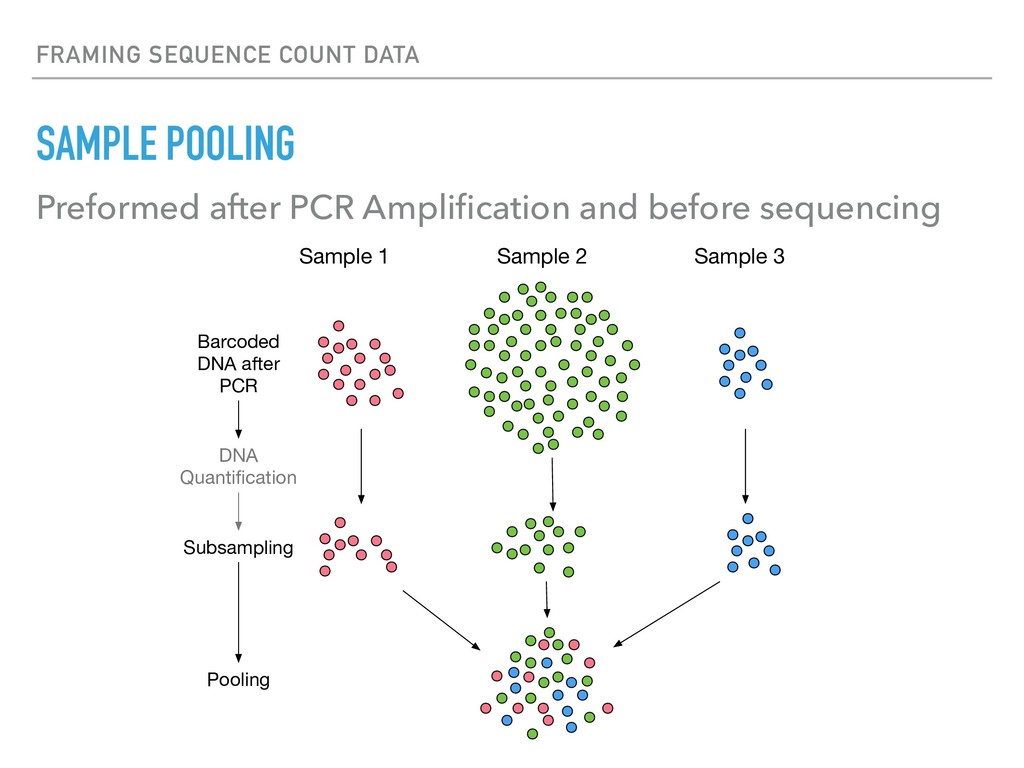
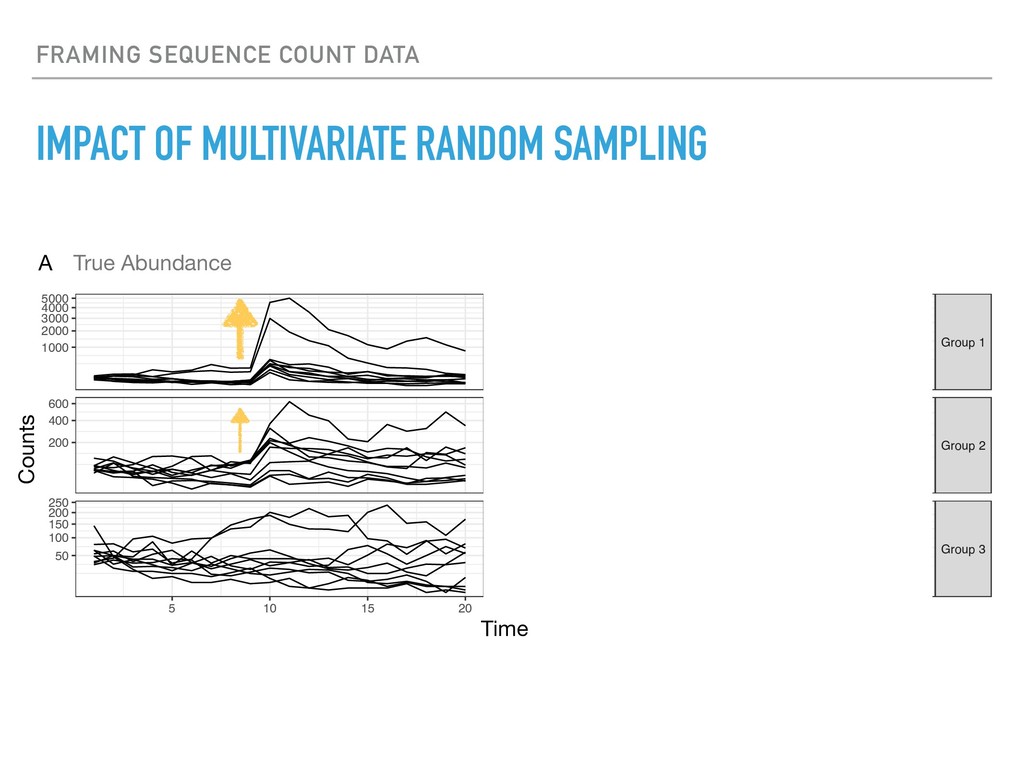
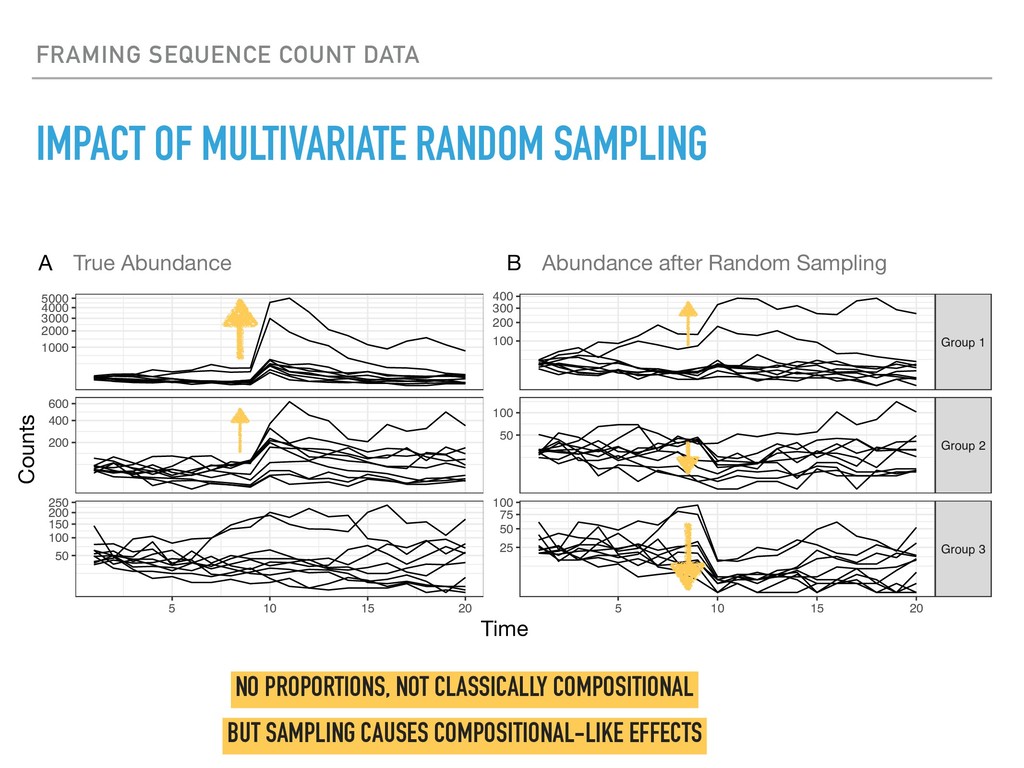
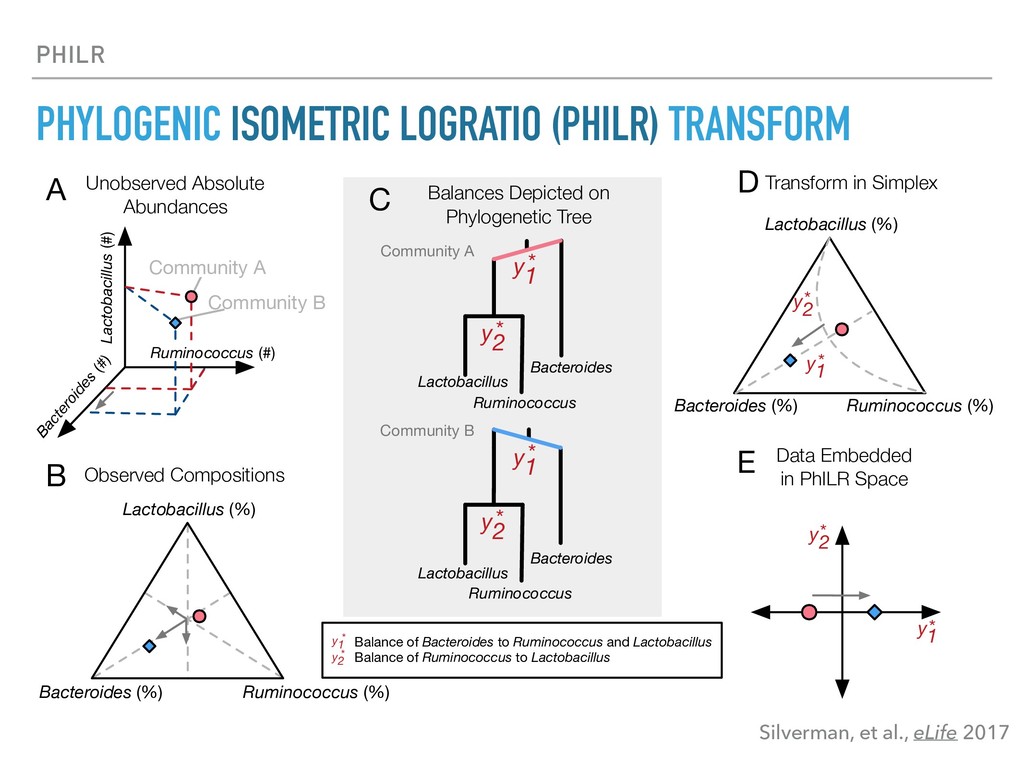
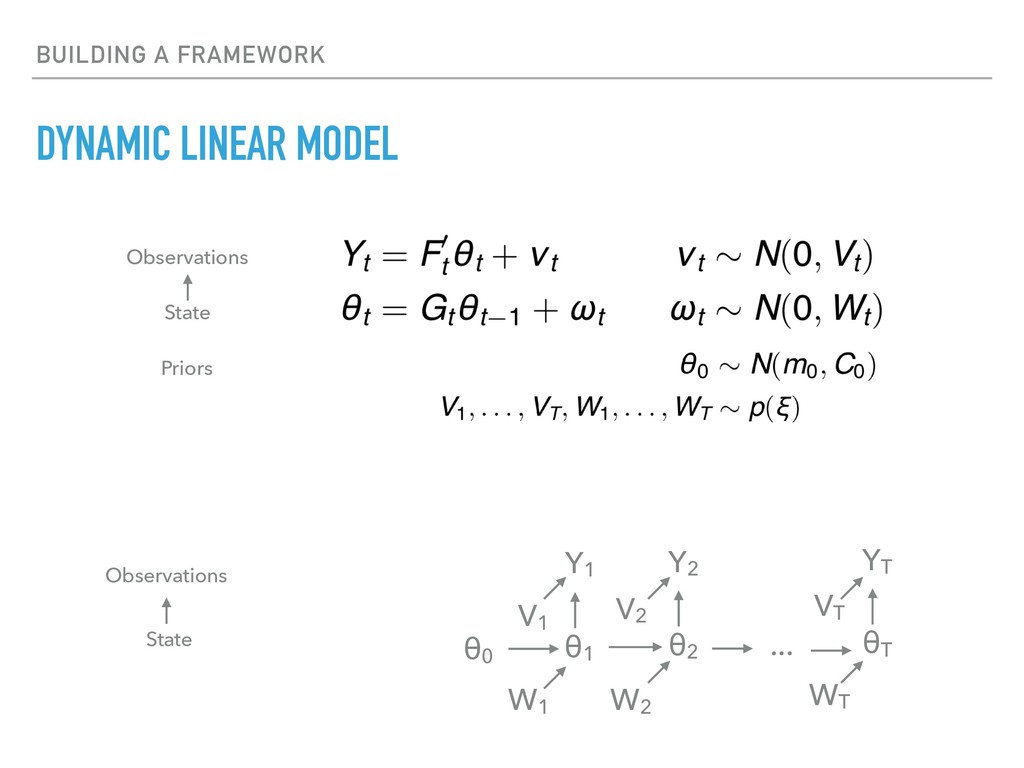
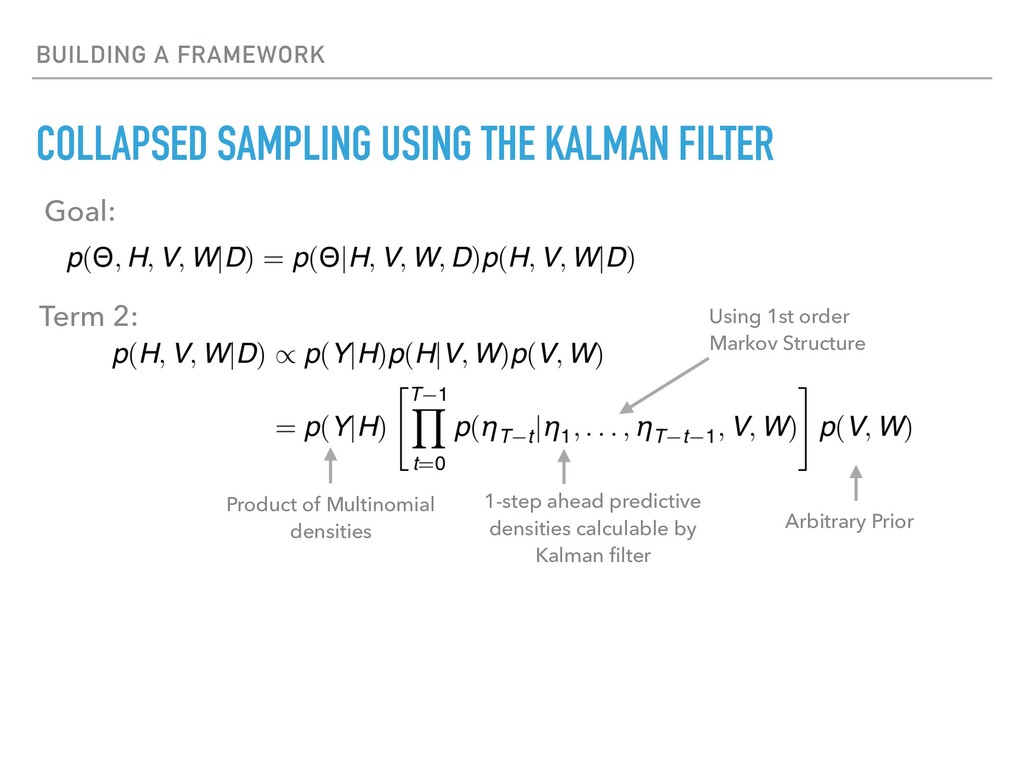
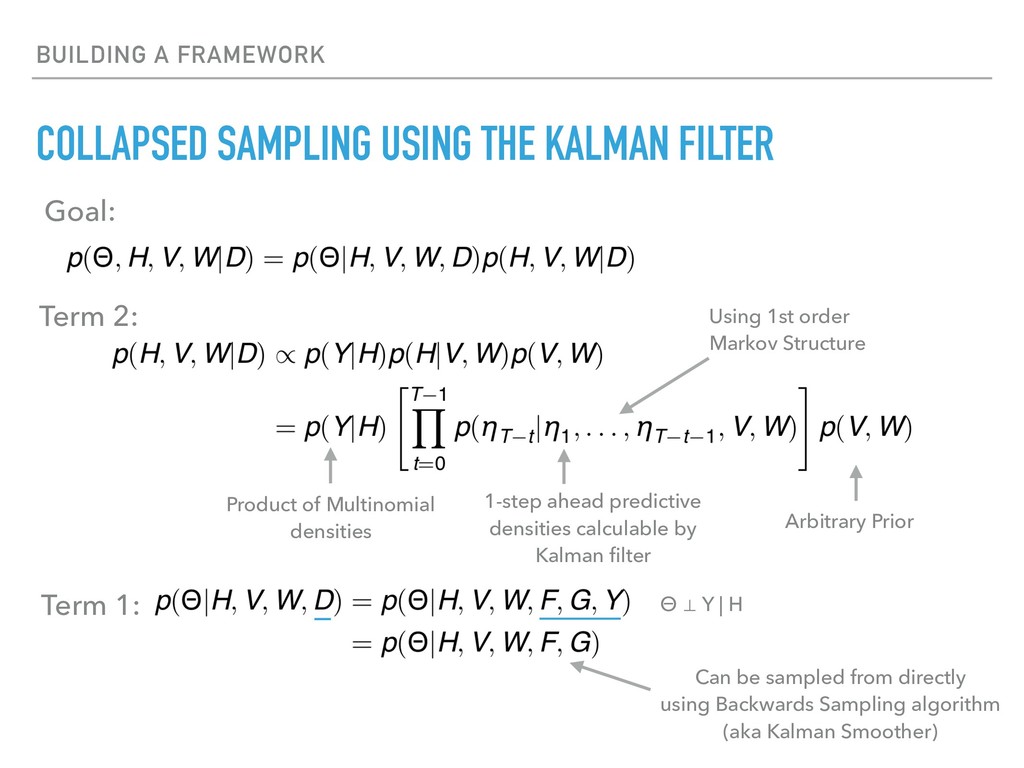
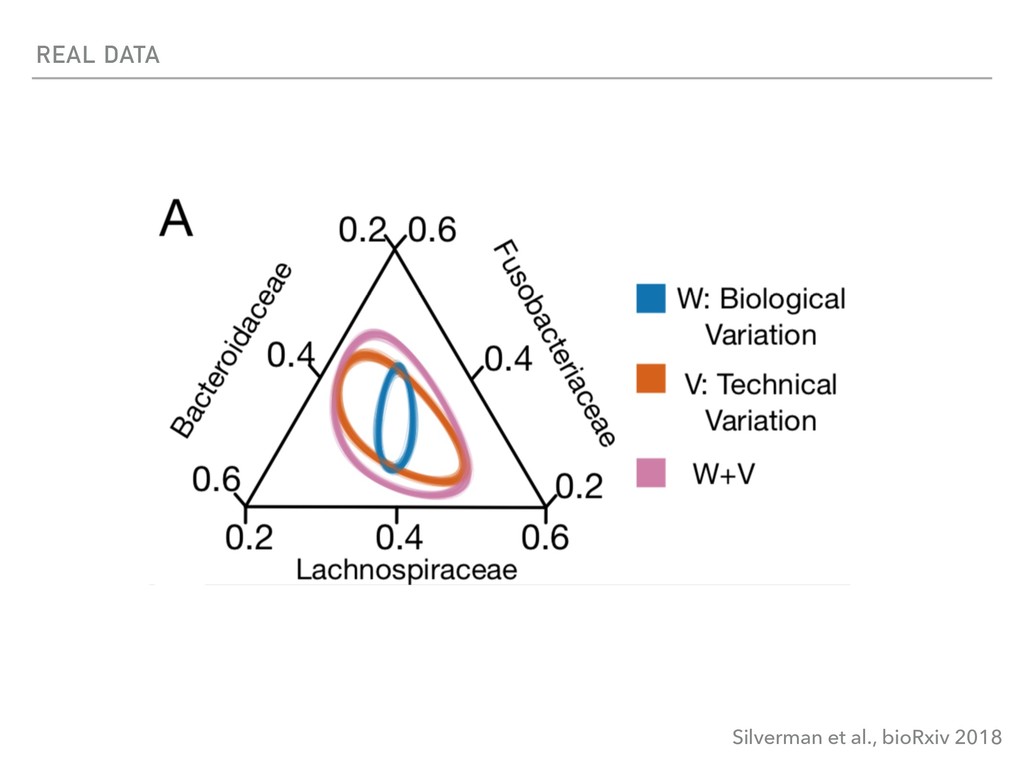
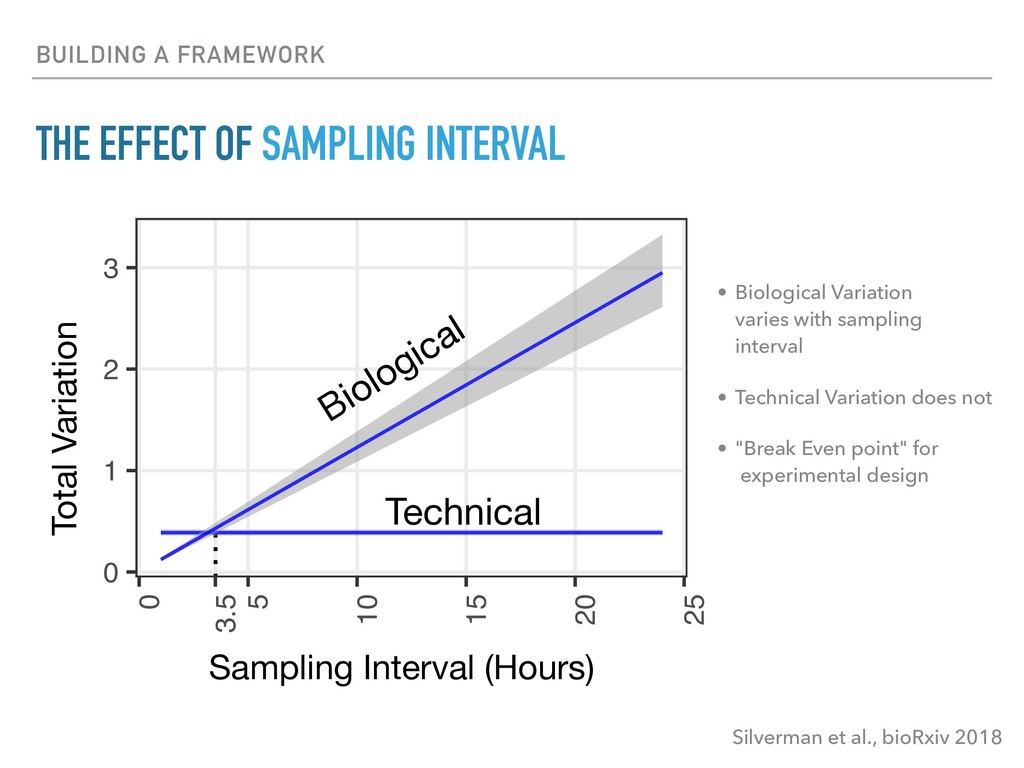
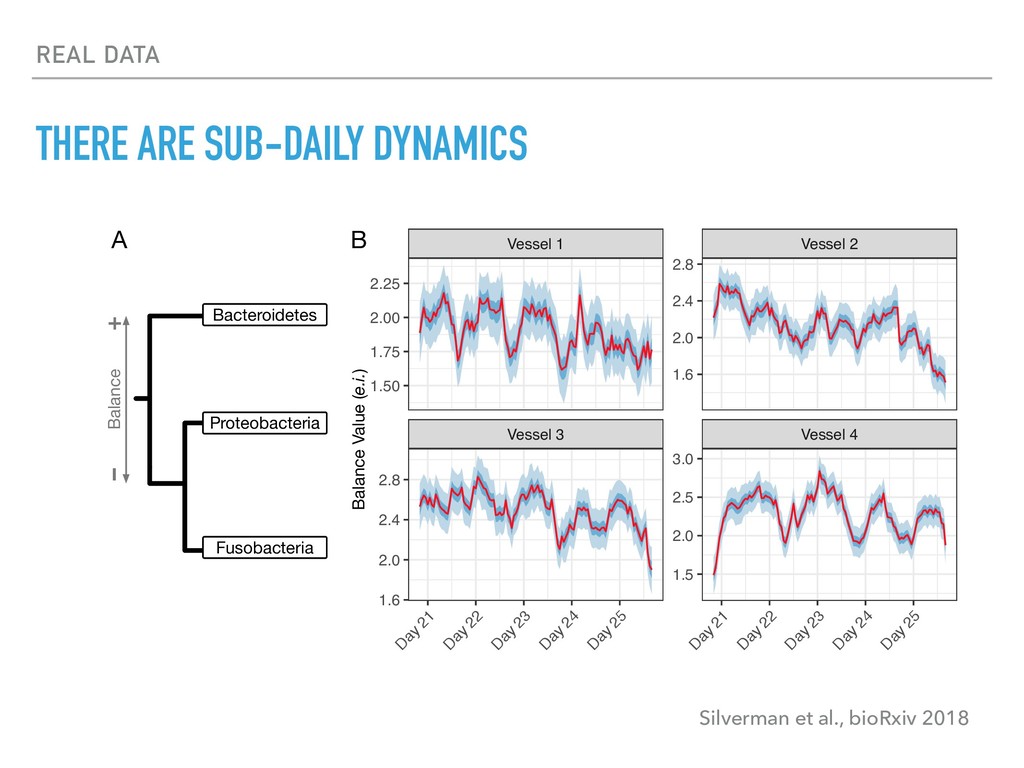
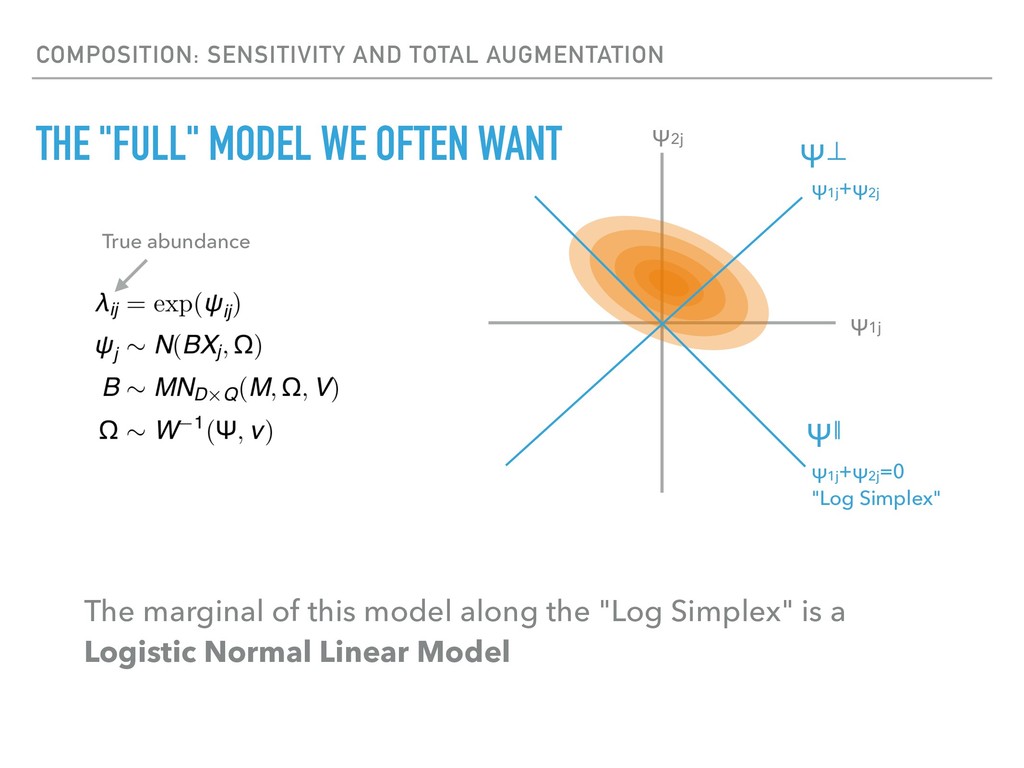
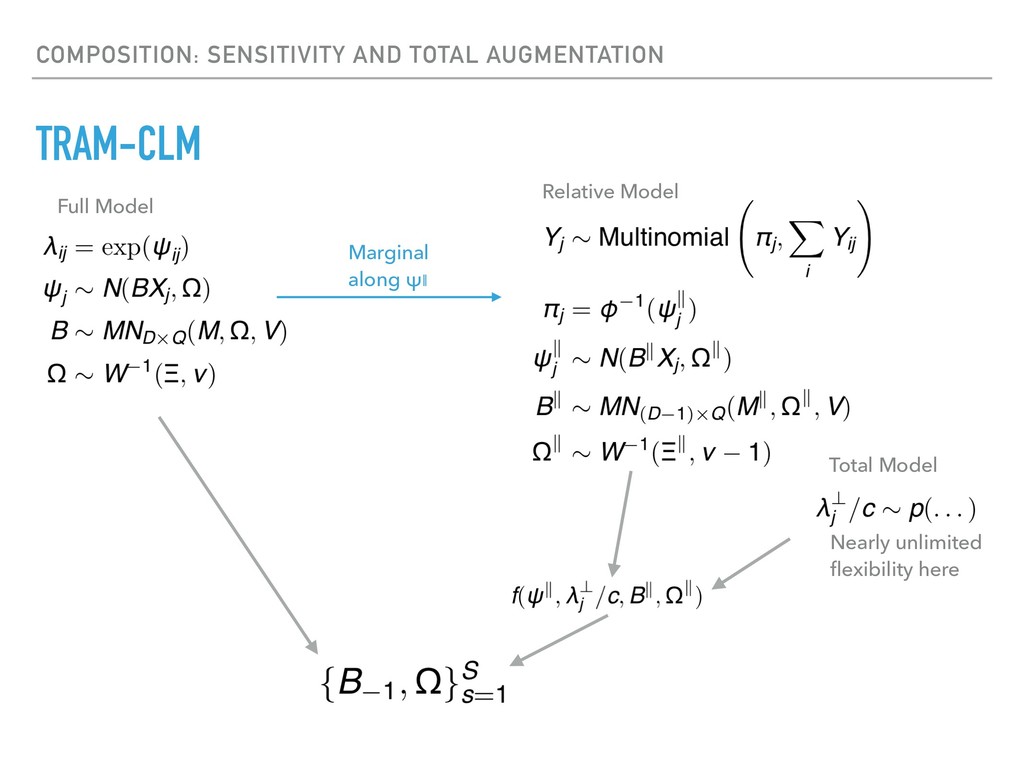
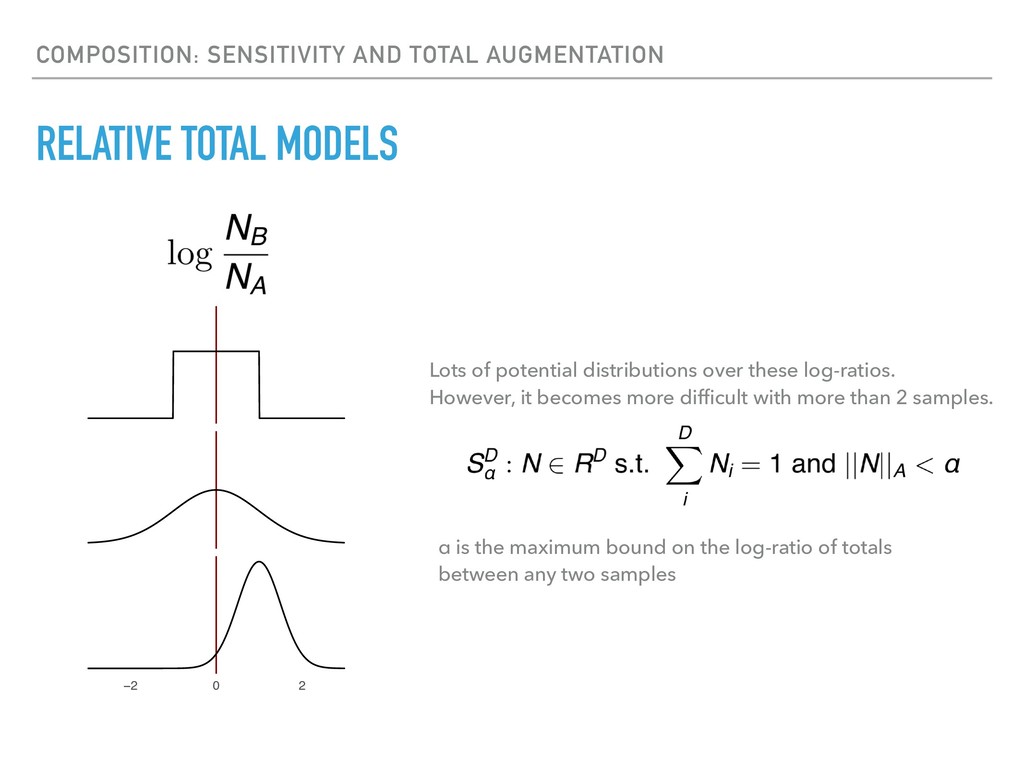
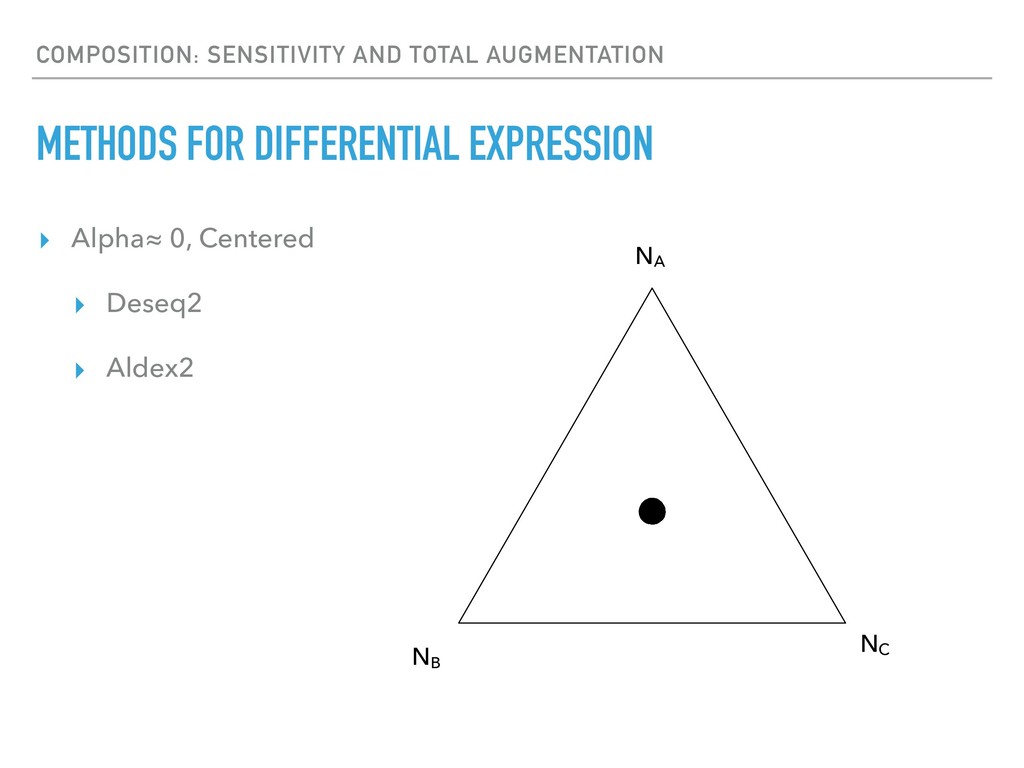
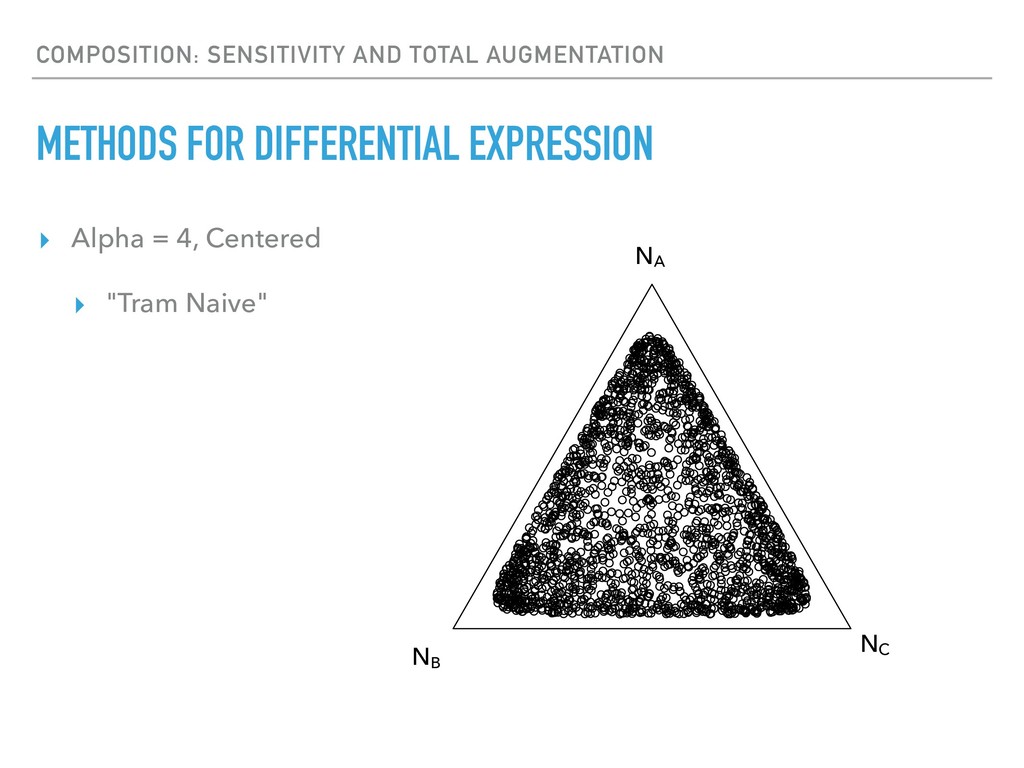
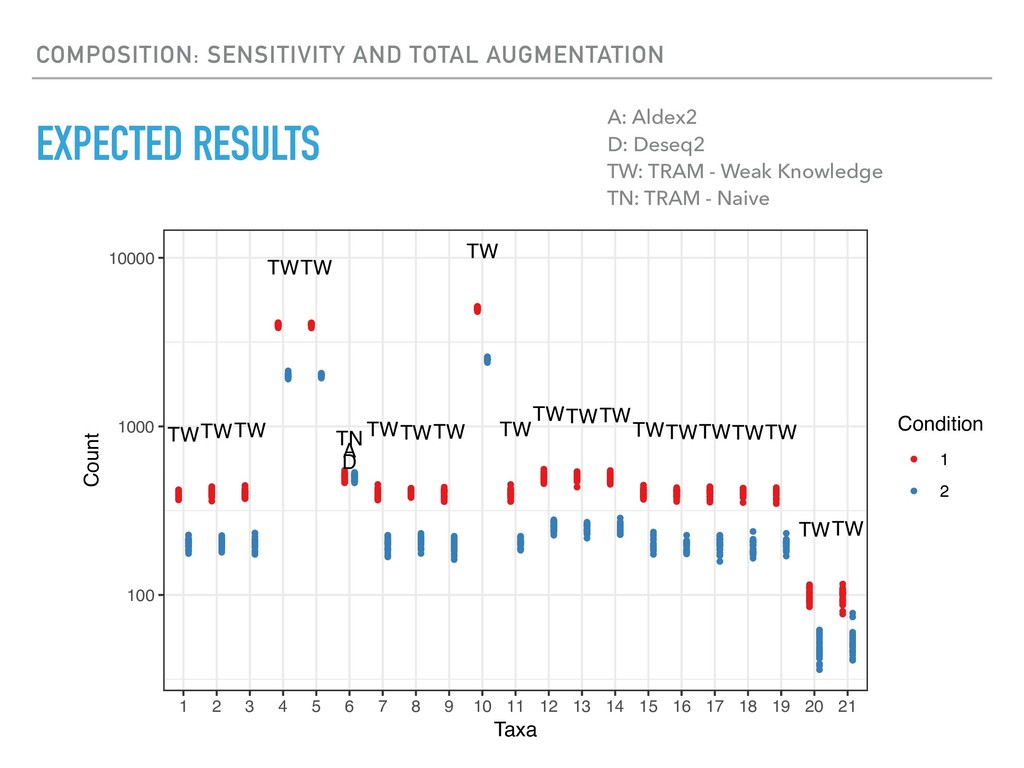
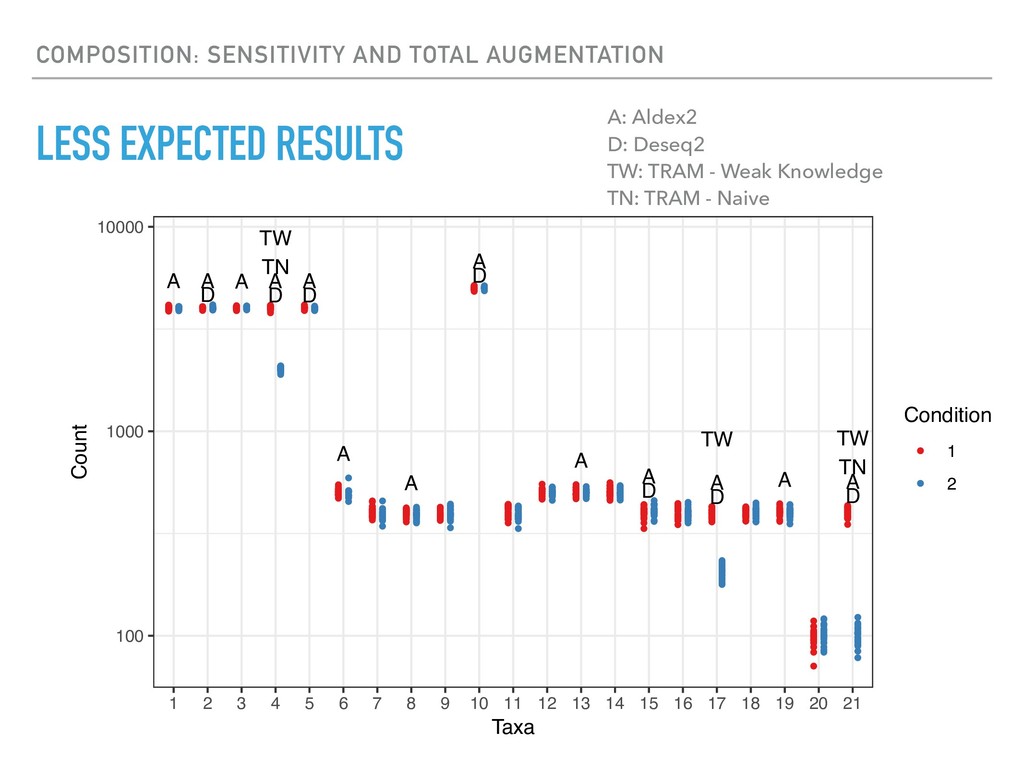
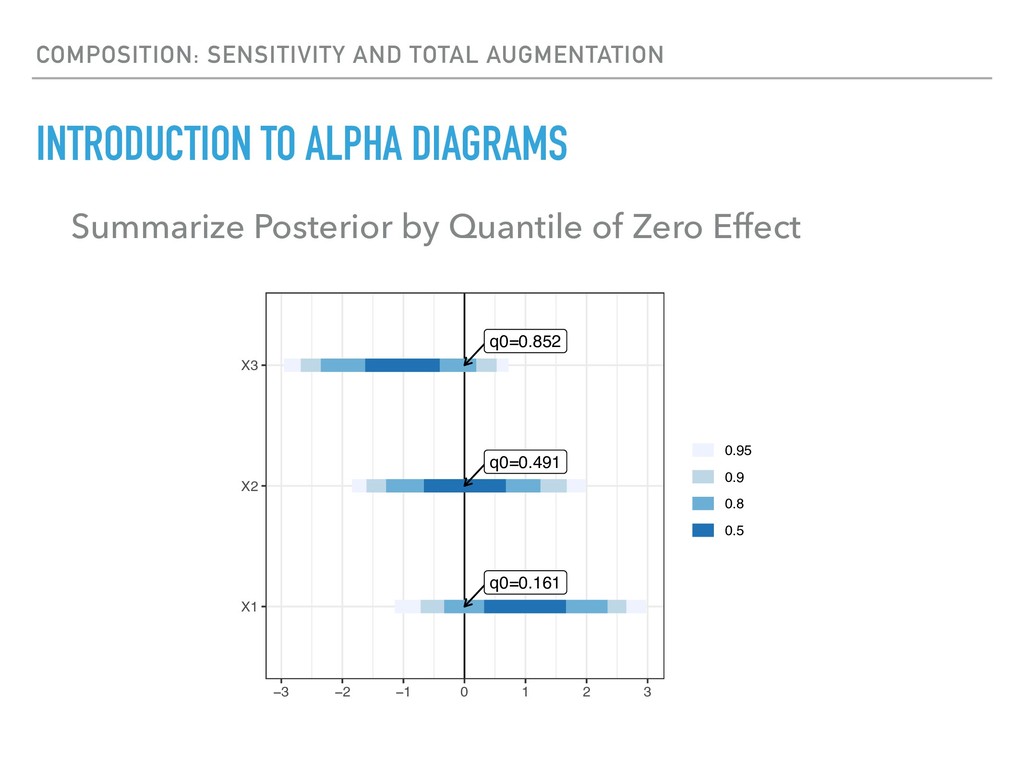
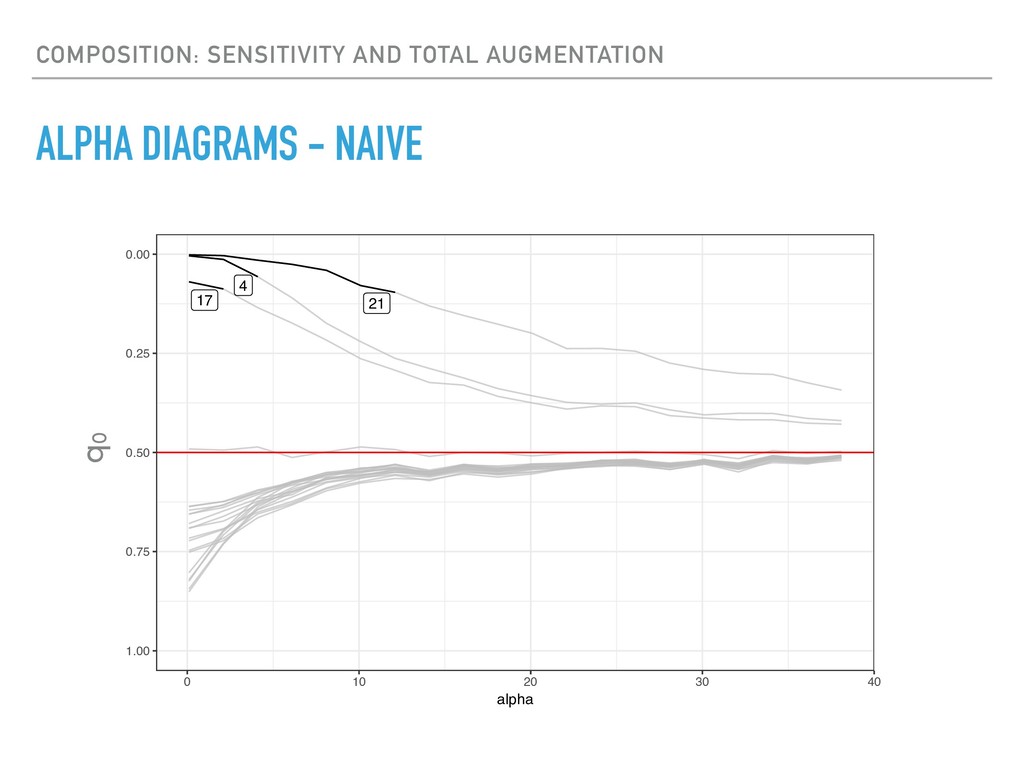
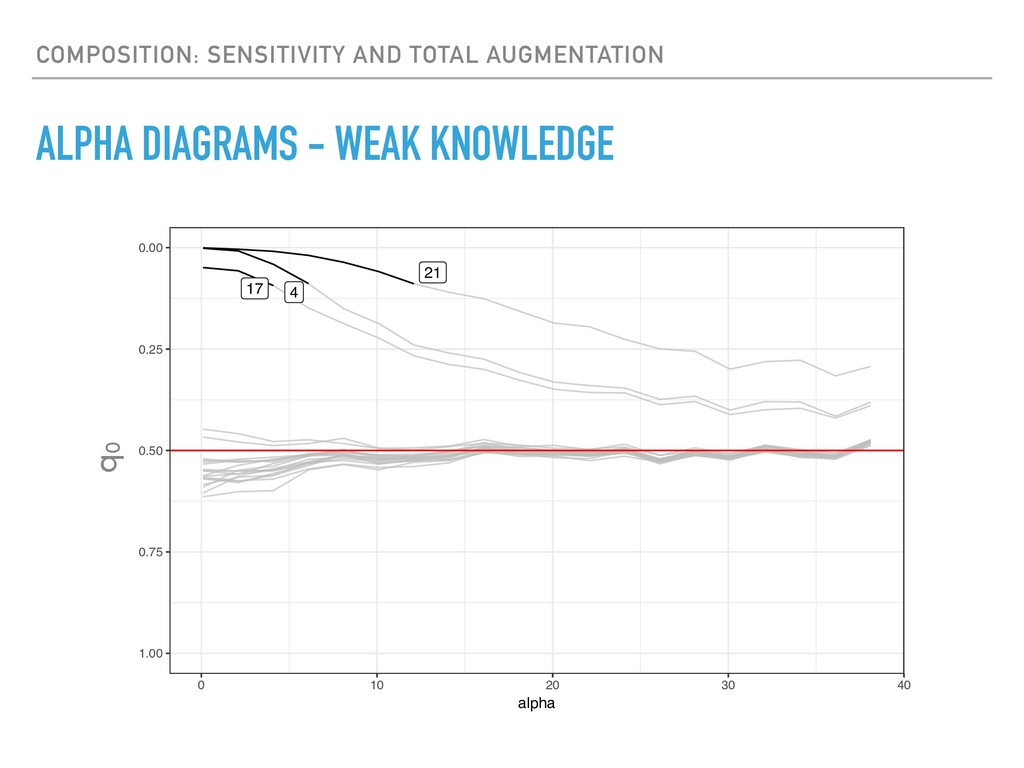
Sequence counting via high-throughput DNA sequencing underlies many studies including 16s rRNA sequencing as well as single-cell or bulk RNA sequencing. However, due to the the measurement process, sequence count data contains information regarding only the relative abundances of sequences. Such relative data, often termed compositional, is known to cause problems in many statistical analyses. Moreover other forms of technical variation and bias complicate analysis of sequence count data. In this talk I will introduce statistical and geometric tools to address these challenges. In particular, by combining these methods with experimental design, I will demonstrate how biological and technical variation in longitudinal studies can be partitioned. Additionally, I will discuss new models that both enable quantification of the sensitivity of real data to compositional effects while also allowing the reconstruction of differential expression and correlation analysis by model augmentation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}