ISMB 2017: Supporting highly scalable scientific data analysis with Galaxy
Technology Talk for ISMB 2017 on 1) Galaxy scalability to thousands of samples, 2) Practical reproducibility with #bioconda, #biocontainers, and virtualization, and 3) [didn't get to this] working with Galaxy entirely from the command line.
design Data collection Data cleaning Data analysis Inference Data Pipeline, inspired by Leek and Peng, Nature 2015 The part we are considering here The part that ends up in the Publication
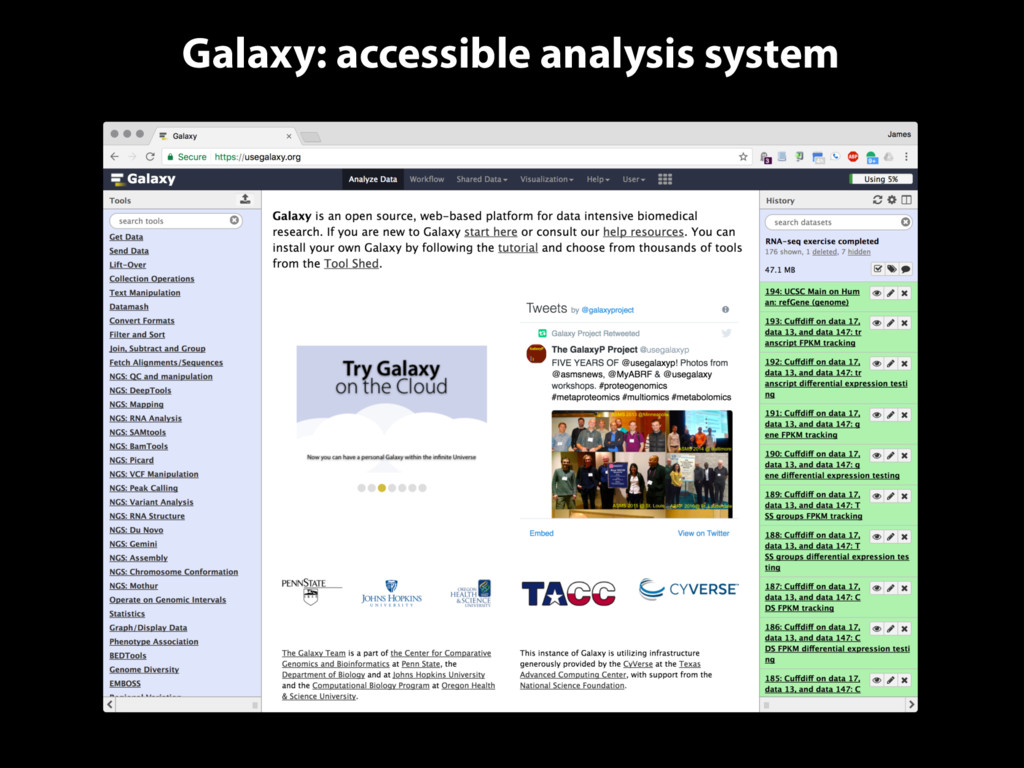
methods, make these methods available to everyone Transparency: Facilitate communication of analyses and results in ways that are easy to understand while providing all details Reproducibility: Ensure that analysis performed in the system can be reproduced precisely and practically
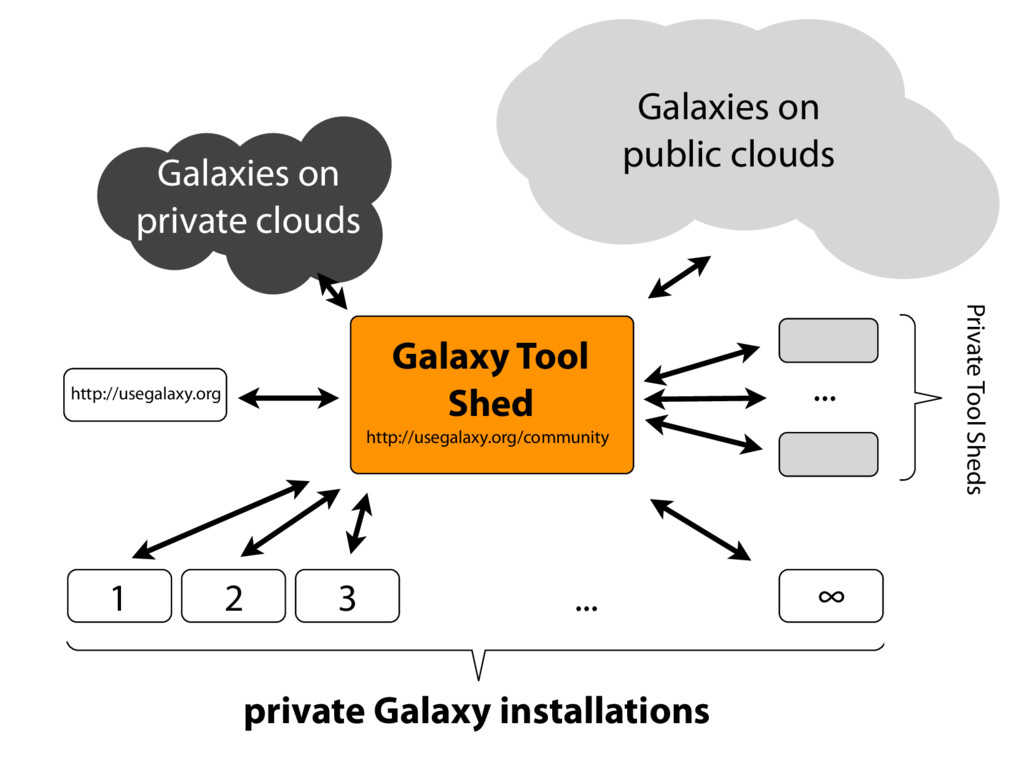
tools, compute resources, terabytes of reference data and permanent storage Open source software that makes integrating your own tools and data and customizing for your own site simple An open extensible platform for sharing tools, datatypes, workflows, ...
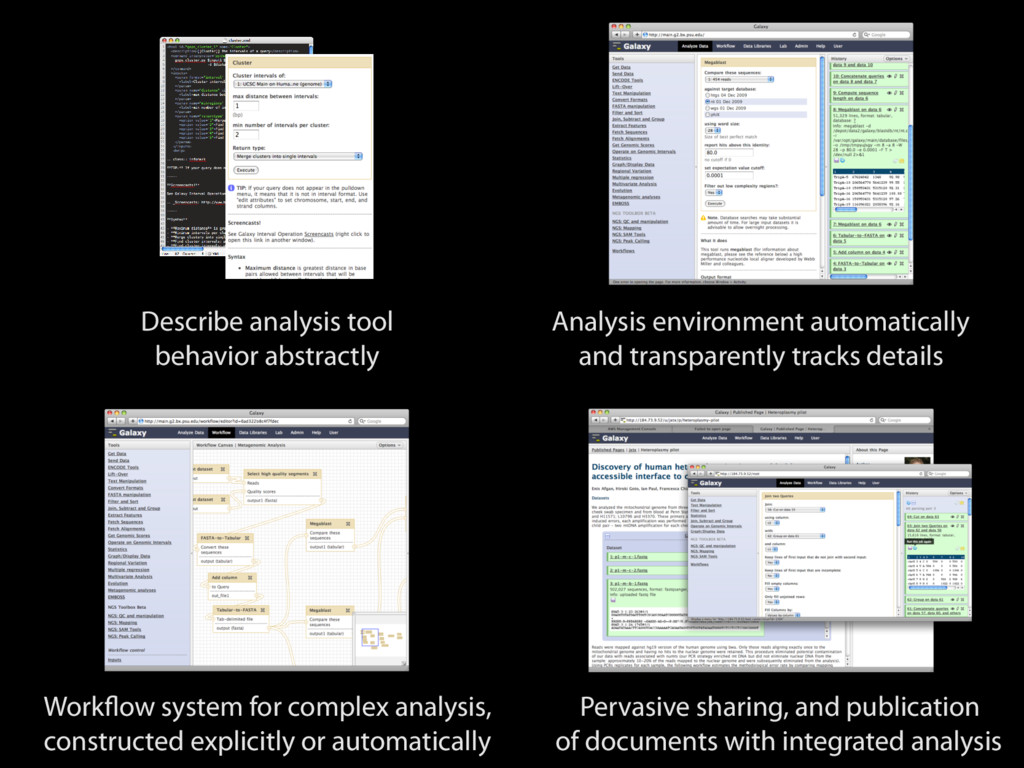
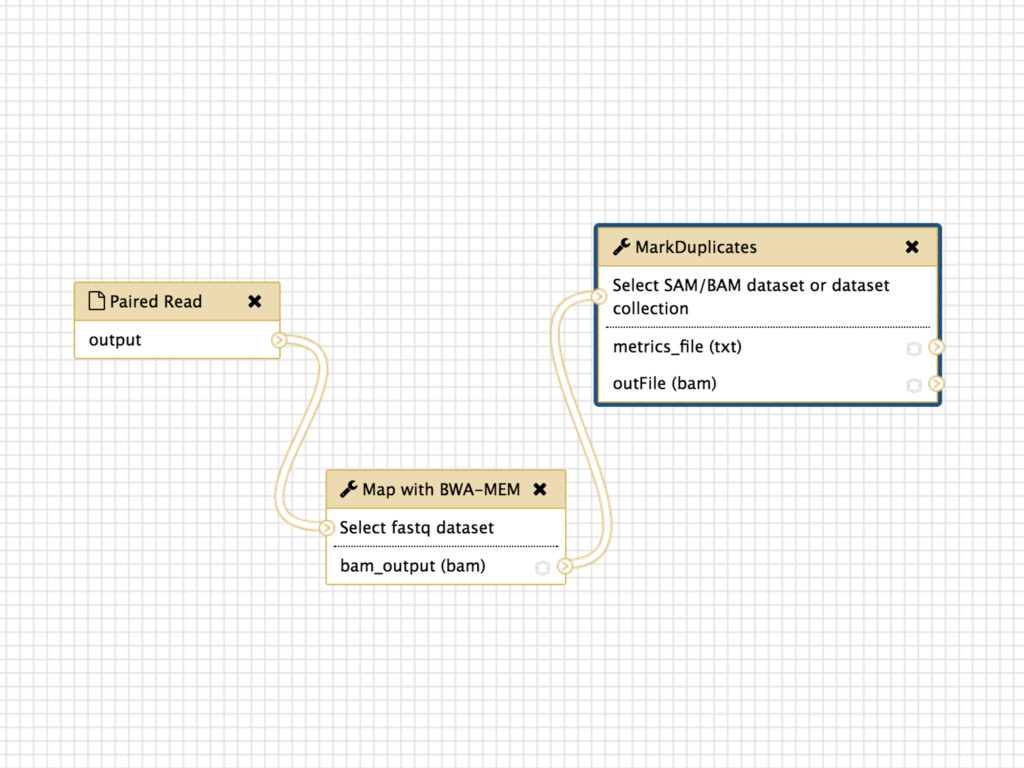
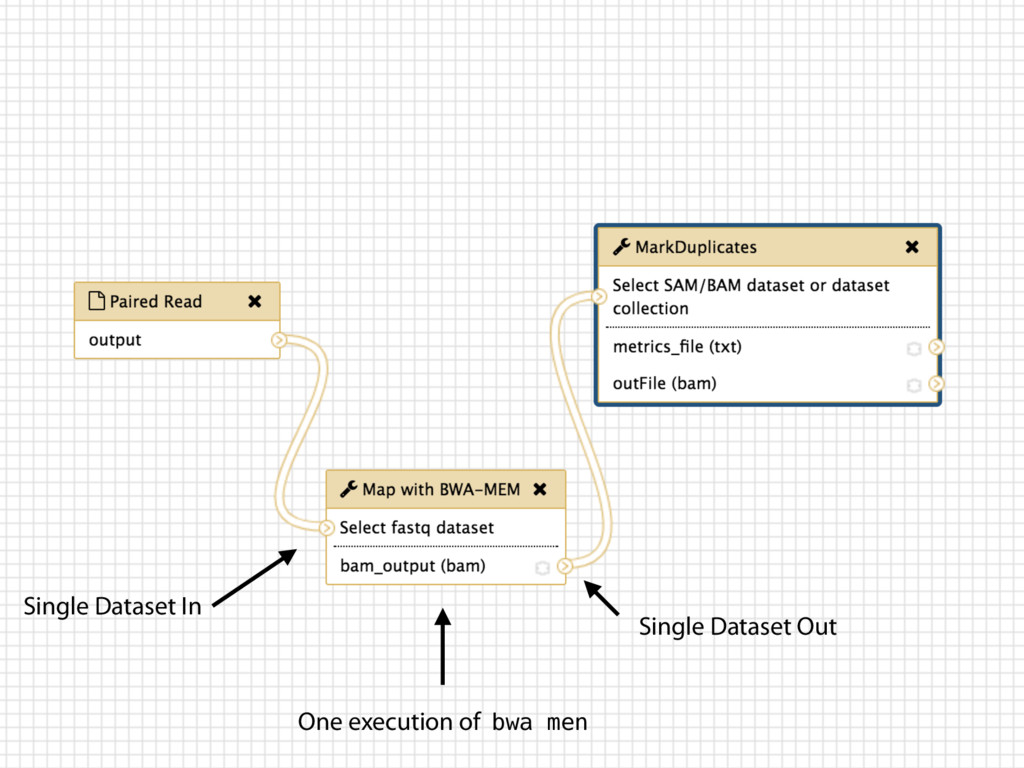
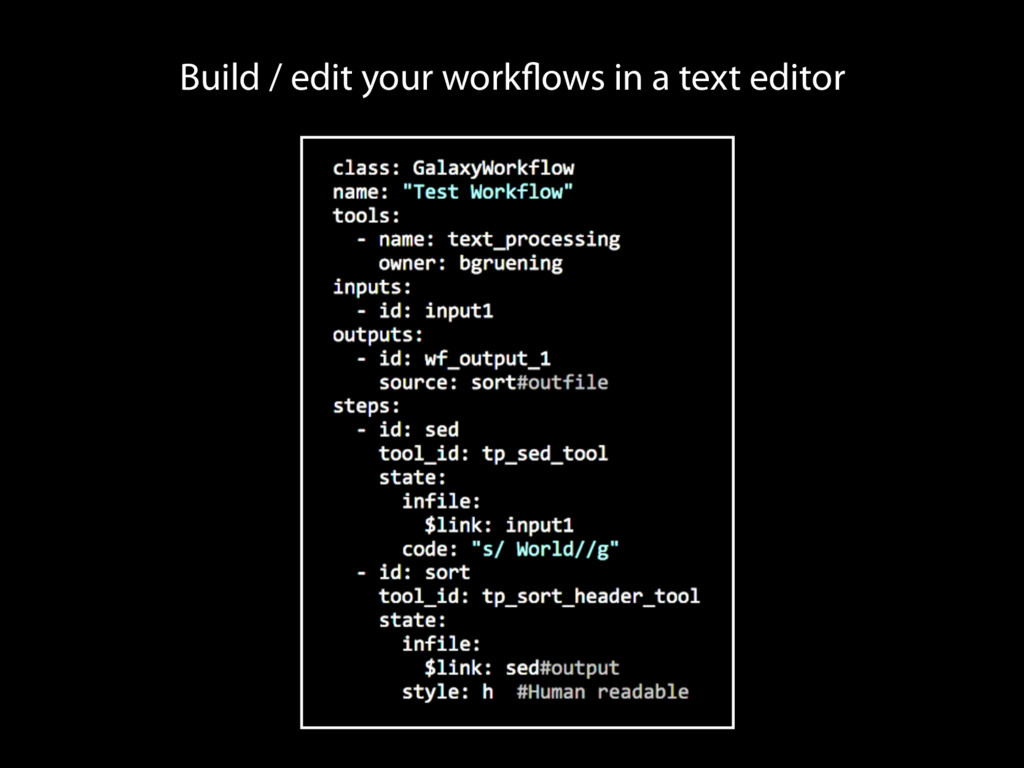
tracks details Workflow system for complex analysis, constructed explicitly or automatically Pervasive sharing, and publication of documents with integrated analysis
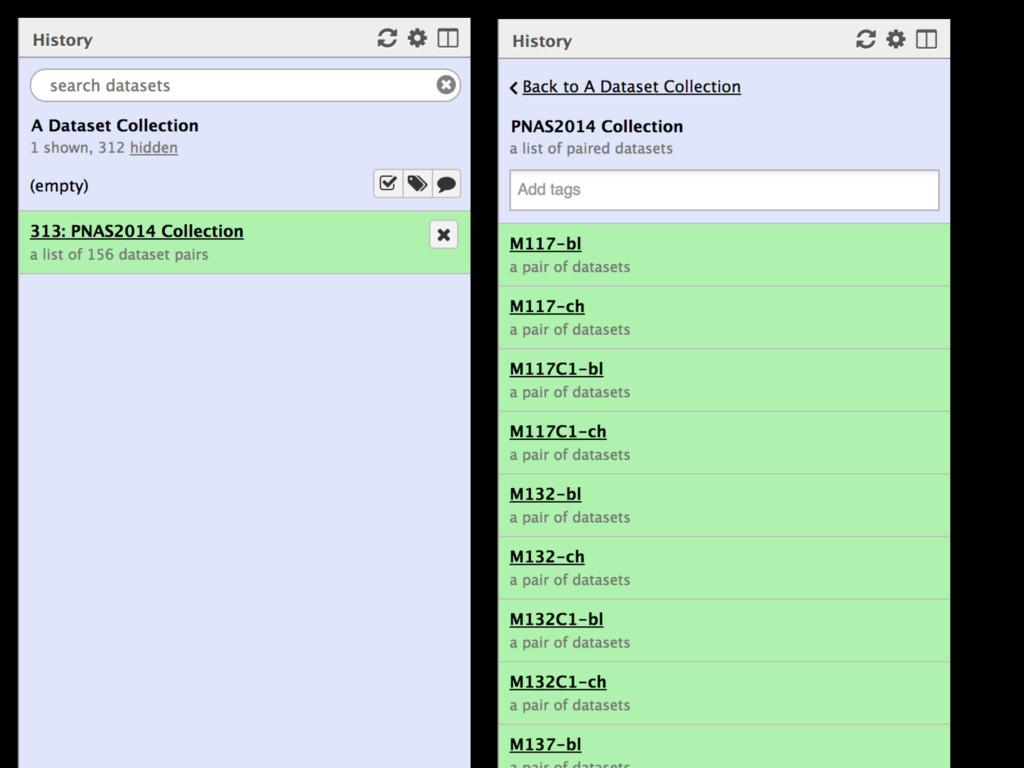
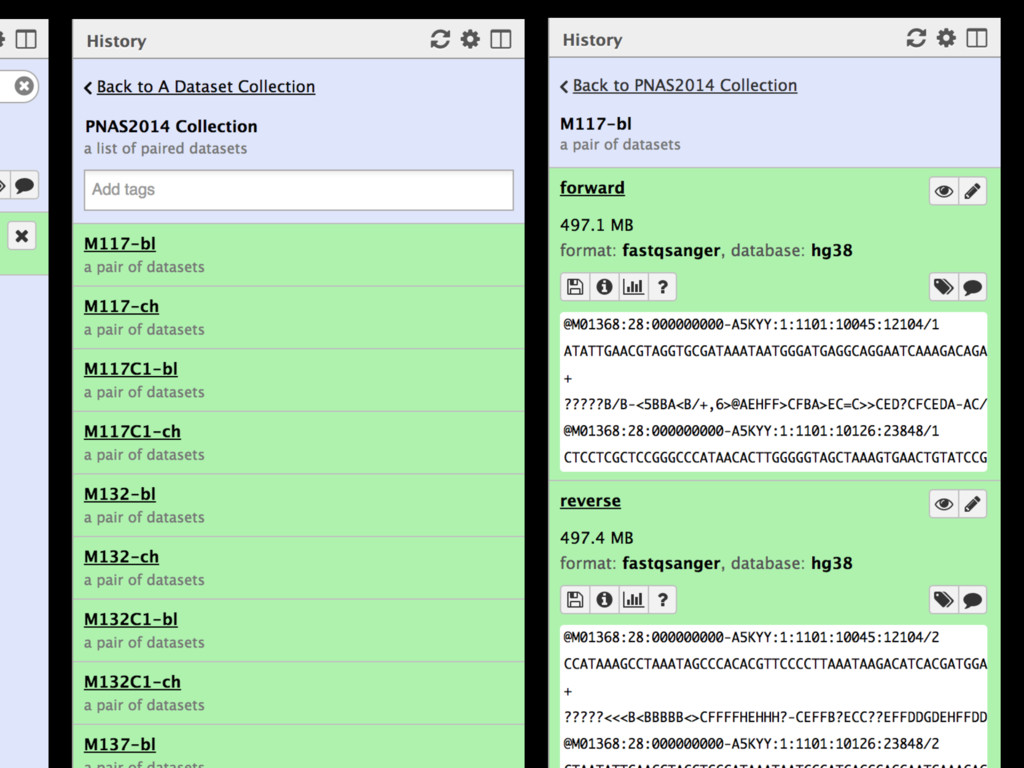
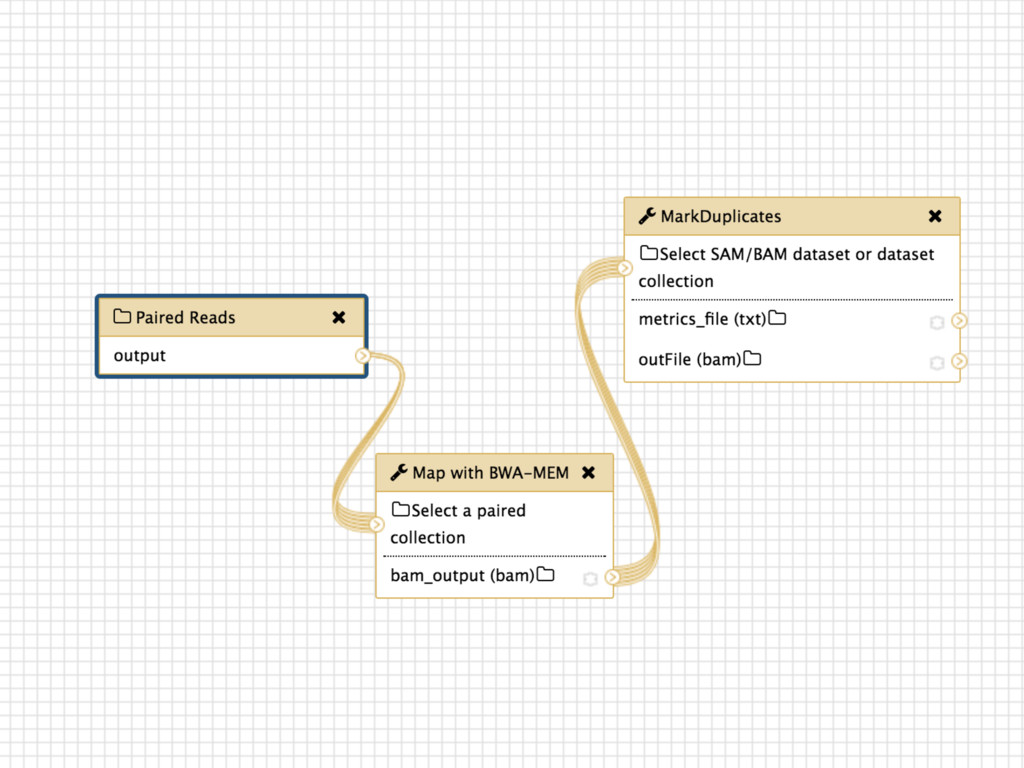
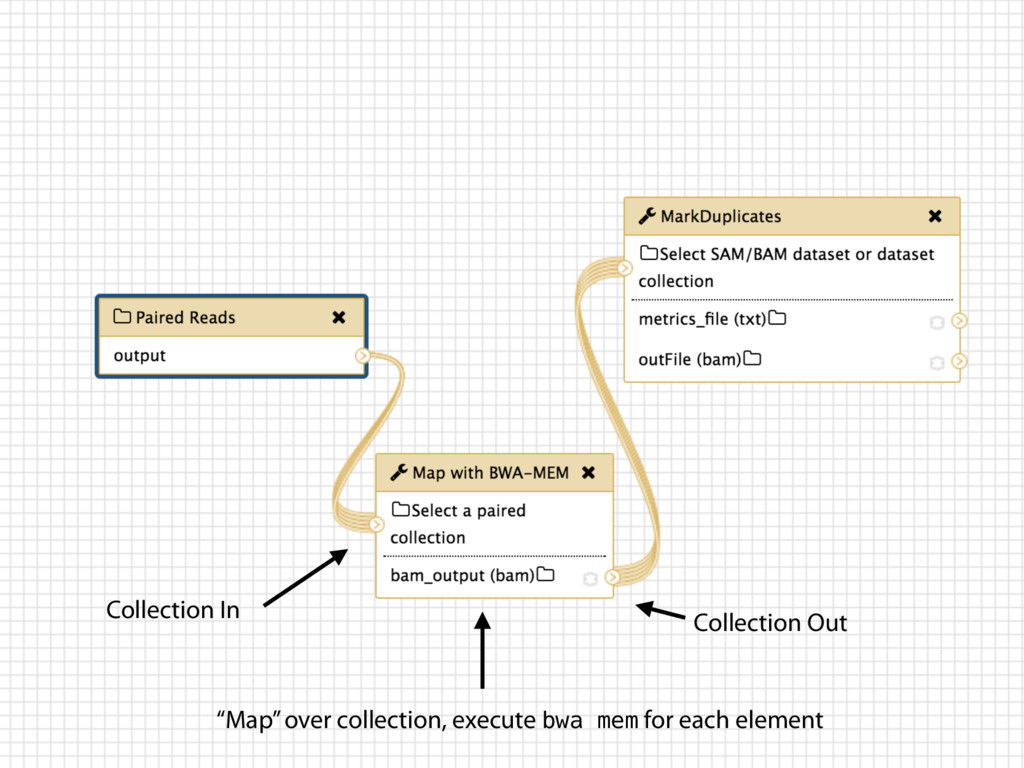
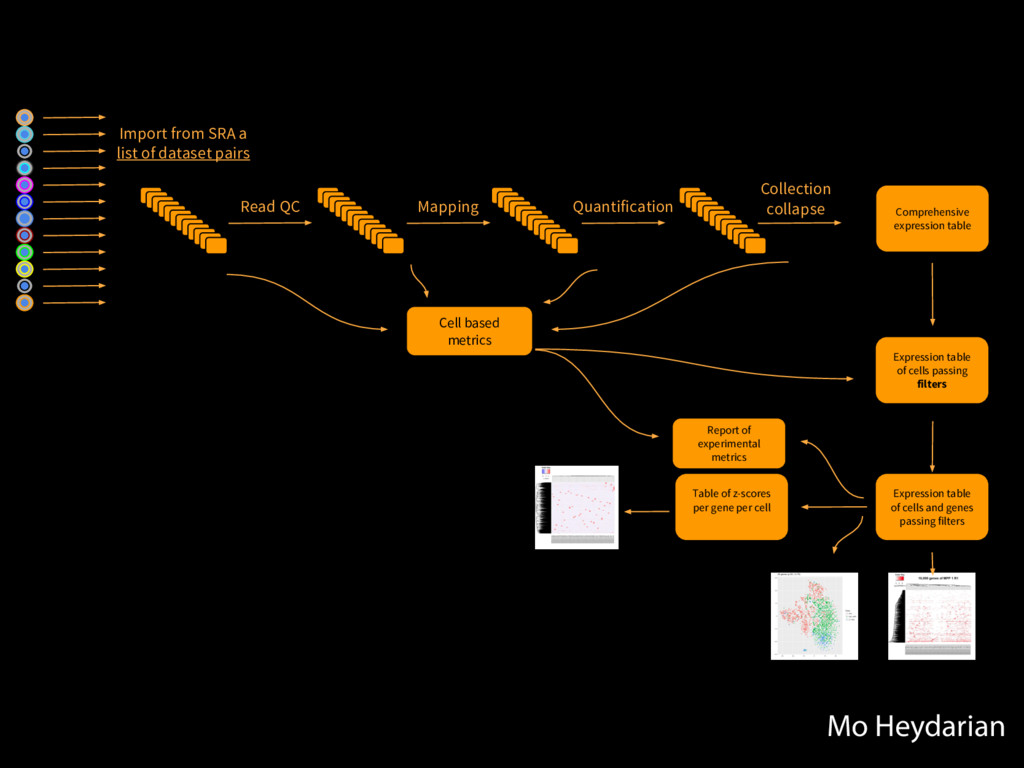
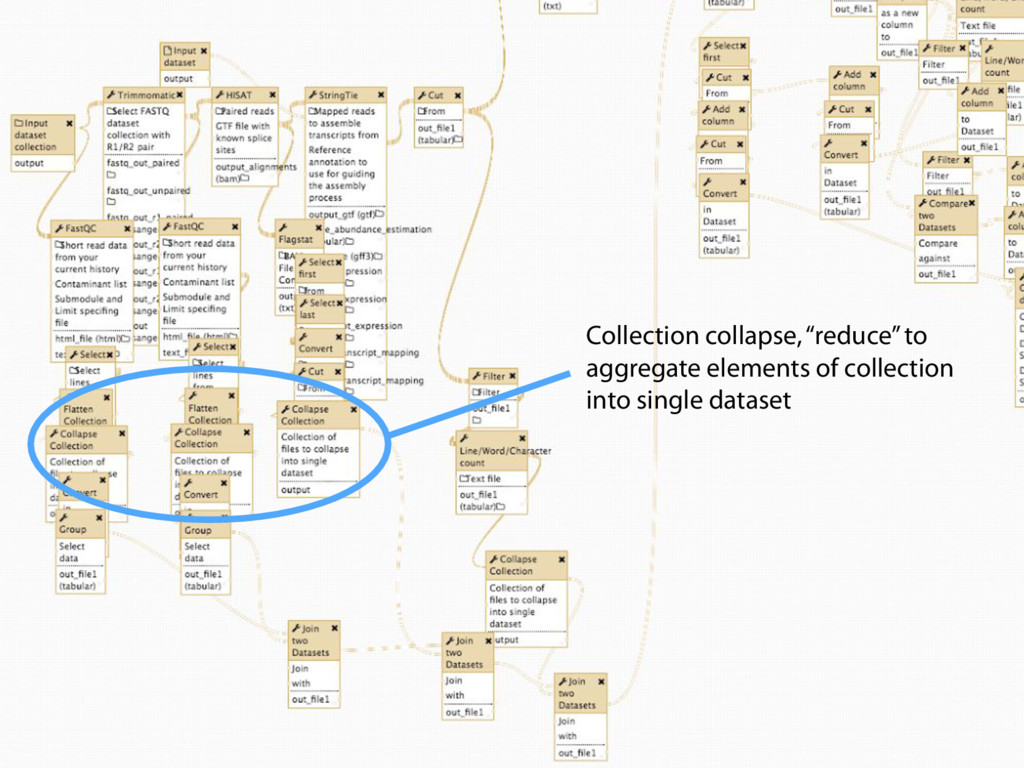
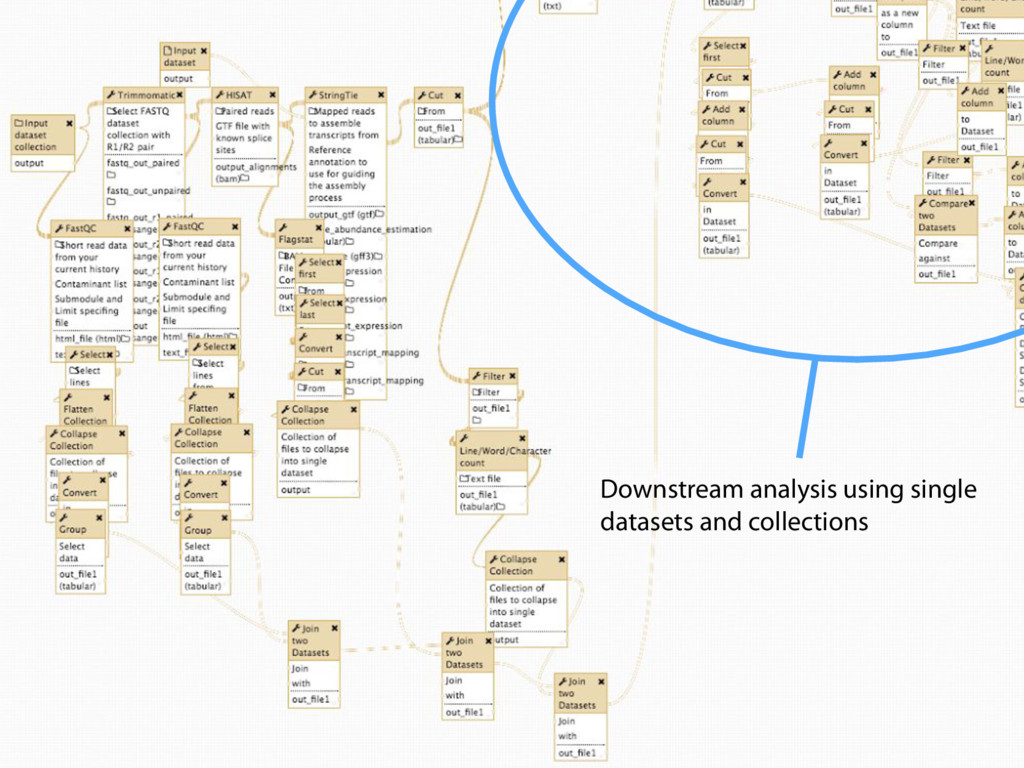
Collections Collapsing single cell data to single tables Collection collapse (“reduce”) Operating on an unknown number of columns Melt and cast tools Visualize hundreds of samples easily New visualization tools
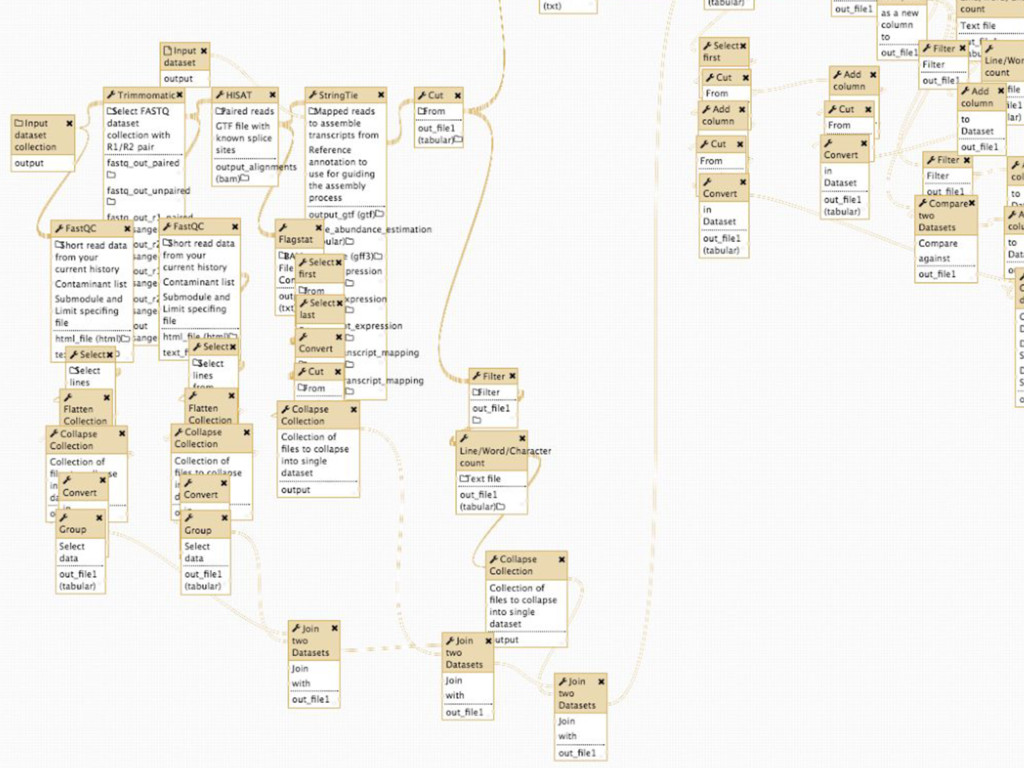
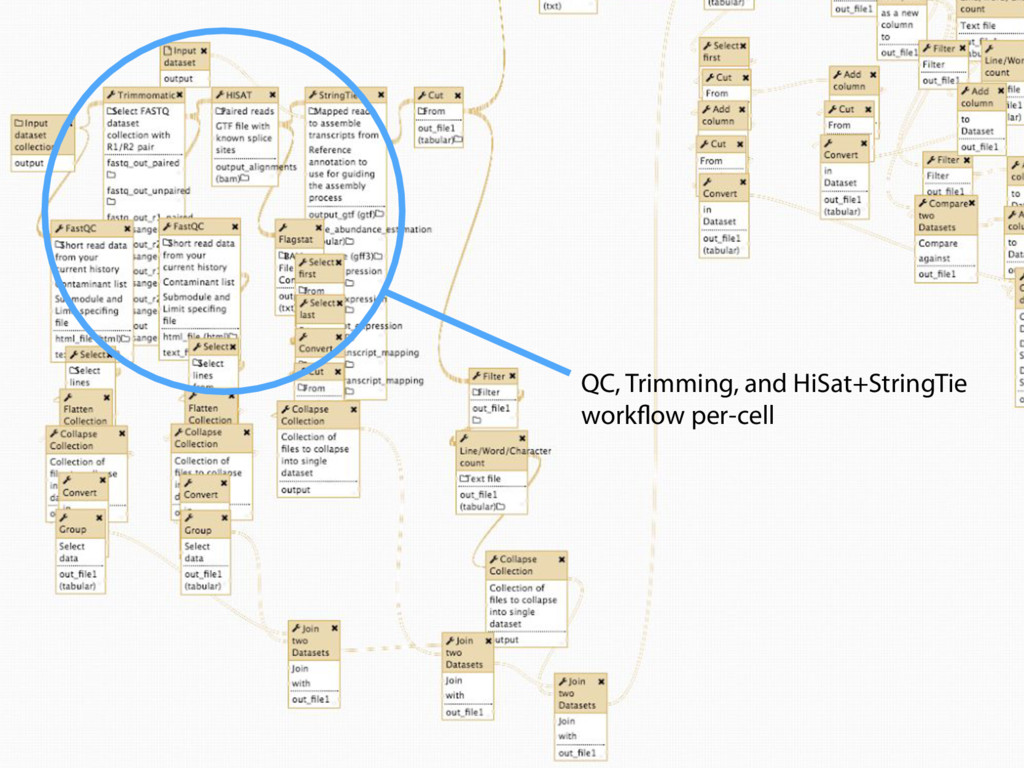
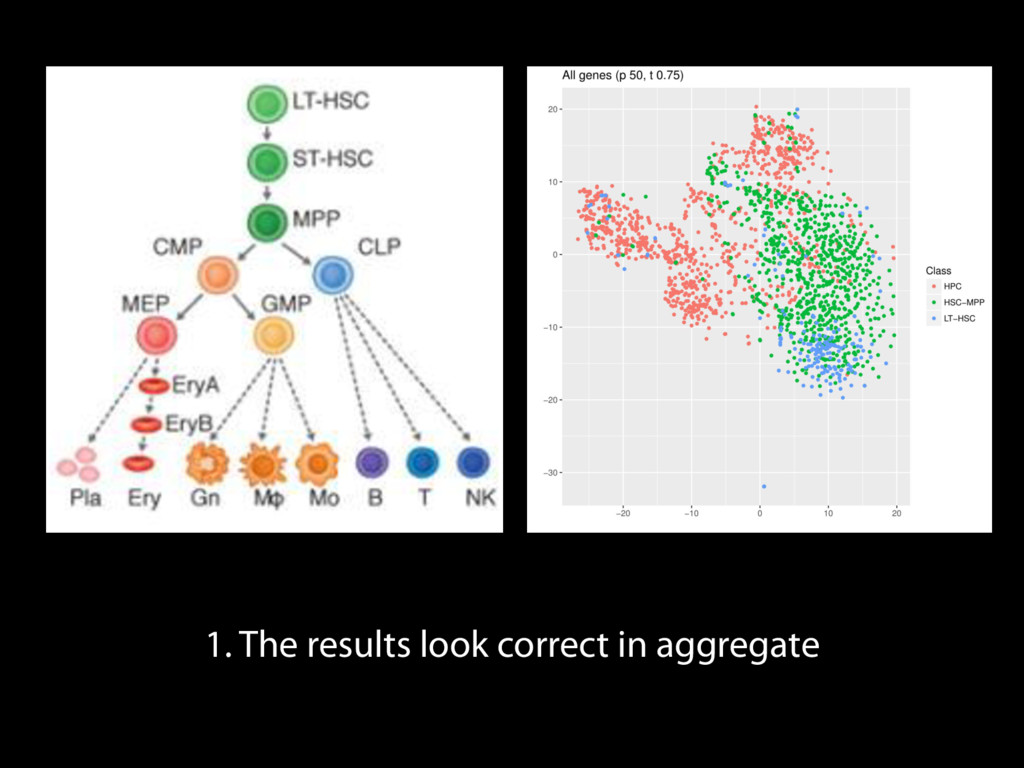
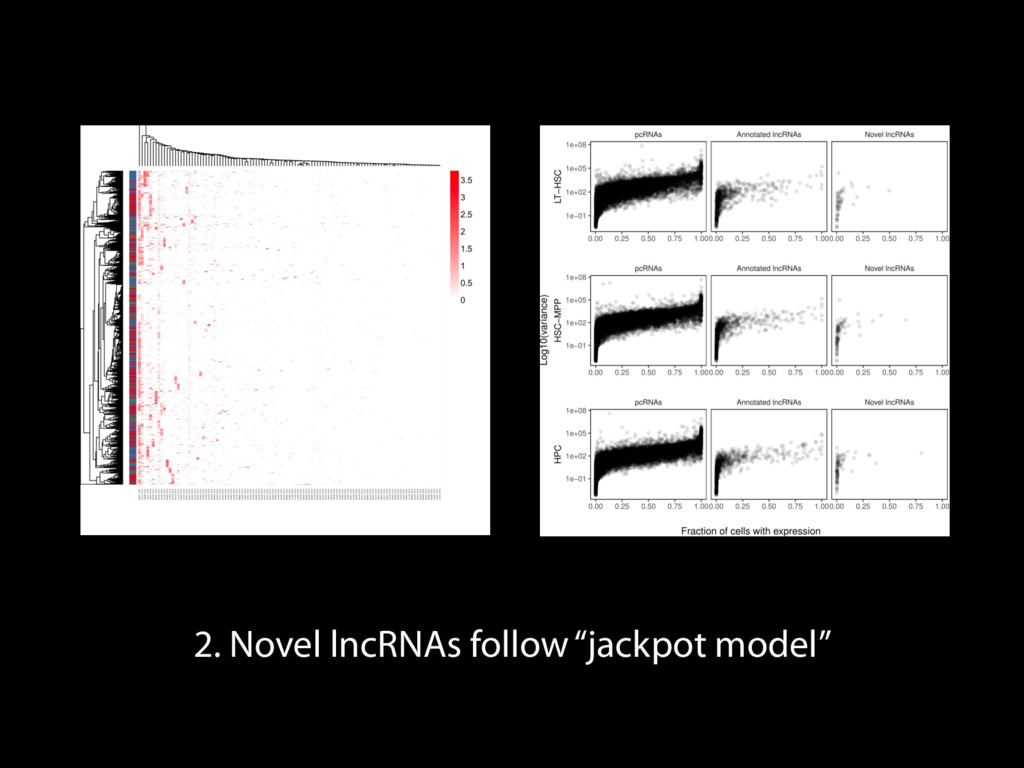
Mapping Quantification Comprehensive expression table Collection collapse Cell based metrics Expression table of cells passing filters Expression table of cells and genes passing filters Table of z-scores per gene per cell Report of experimental metrics Mo Heydarian
108 h generated 100,149 history items!! Zero errors! Big Fella taking big strides Processing all 3840 cells took 108 hours and generated 100,149 history items!!! Zero errors! 3,840 cells: 108 hours and 100,149 history items. Zero errors. Mo Heydarian
tools Community response has been phenomenal However, packaging is challenging — it never ends! Need to move to a model that pulls in and integrates with a broader community
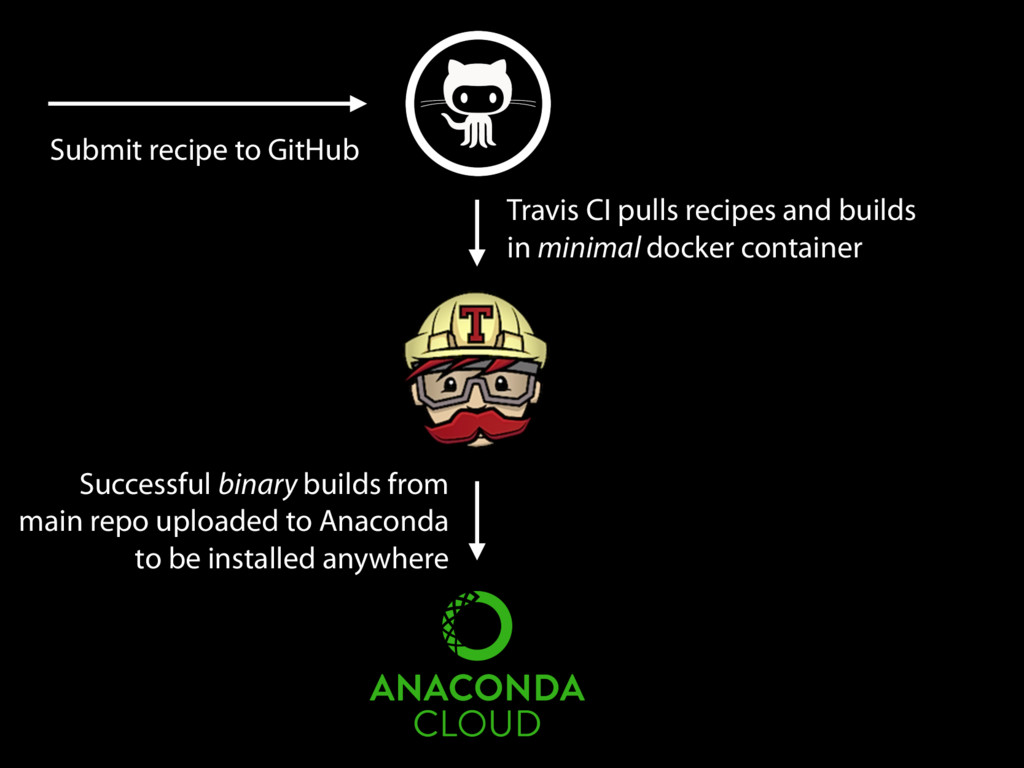
of software packages and their dependencies and switching easily between them” ~2200 recipes for software packages (as of yesterday) All packages are automatically built in a minimal environment to ensure isolation and portability
the kernel level up Containers — lightweight environments with isolation enforced at the OS level, complete control over all software Adds a complete ecosystem for sharing, versioning, managing containers — e.g. Docker hub, quay.io
— analysis are isolated and environment is the same every time Archive that container — containers are lightweight thanks to layers — and the analysis can always be recreated
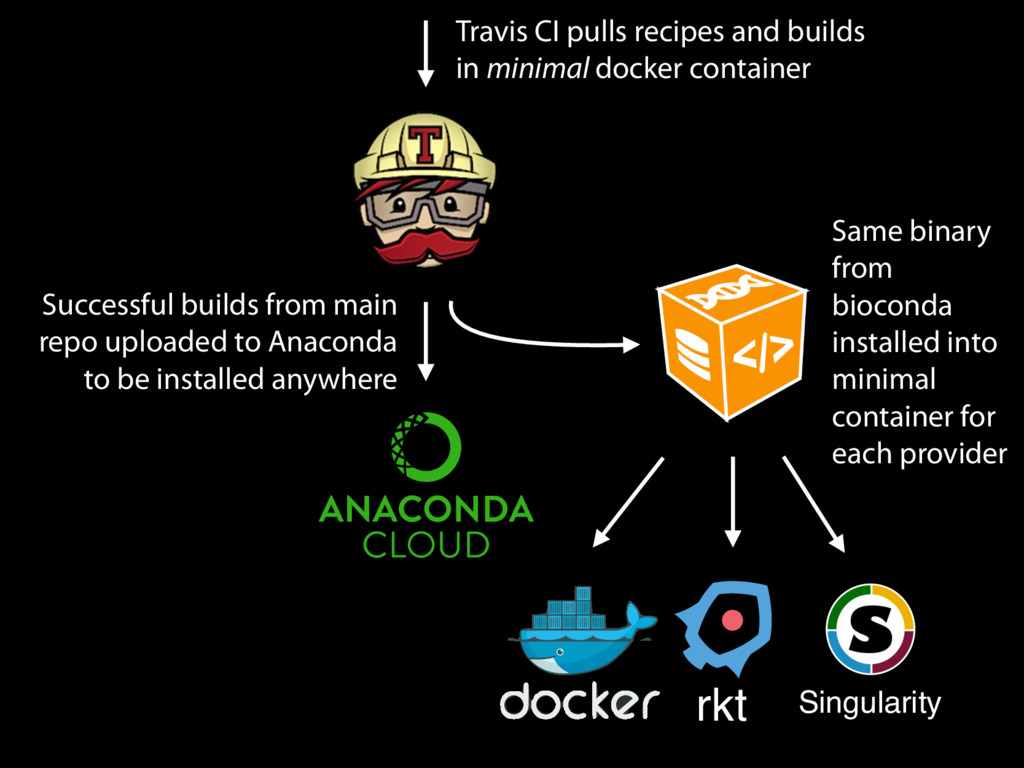
in Conda/ Bioconda, we can build a container with just that software on a minimal base image If we use the same base image, we can reconstruct exactly the same container (since we archive all binary builds of all versions) With automation, these containers can be built automatically for every package with no manual modification or intervention (e.g. mulled)
Successful builds from main repo uploaded to Anaconda to be installed anywhere Same binary from bioconda installed into minimal container for each provider rkt Singularity
Galaxy is configured this can be resolved with conda, with biocontainers… …or environment modules, or brew, guix, … (Resolvers are completely pluggable)
Bouvier, Martin Cěch, John Chilton, Dave Clements, Nate Coraor, Carl Eberhard, Jeremy Goecks, Björn Grüning, Sam Guerler, Mo Heydarian, Jennifer Hillman-Jackson, Anton Nekrutenko, Eric Rasche, Nicola Soranzo, Marius van den Beek BioConda and Biocontainers: Johannes Köster, Ryan Dale, Björn Grüning, … All contributors to and users of all of the projects I’ve talked about NHGRI (HG005133, HG004909, HG005542, HG005573, HG006620) NIDDK (DK065806) and NSF (DBI 0543285, DBI 0850103)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}