Martin Čech, John Chilton, Dave Clements, Nate Coraor, Jeremy Goecks, Björn Grüning, Sam Guerler, Jennifer Hillman-Jackson, Anton Nekrutenko, Helena Rasche, Nicola Soranzo, Marius van den Beek +CloudLaunch+Kubernetes: Nuwan Goonasekera, Pablo Moreno Other lab members : Boris Brenerman, Min Hyung Cho, Peter DeFord, Max Echterling, Nathan Roach, Michael E. G. Sauria, German Uritskiy Collaborators:
Craig Stewart and the group
Ross Hardison and the VISION group
Victor Corces (Emory), Karen Reddy (JHU)
Johnston, Kim, Hilser, and DiRuggiero labs (JHU Biology)
Battle, Goff, Langmead, Leek, Schatz, Timp labs (JHU Genomics) NHGRI (HG005133, HG004909, HG005542, HG005573, HG006620)
NIDDK (DK065806) and NSF (DBI 0543285, DBI 0850103) funded by the National Science Foundation Award #ACI-1445604
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
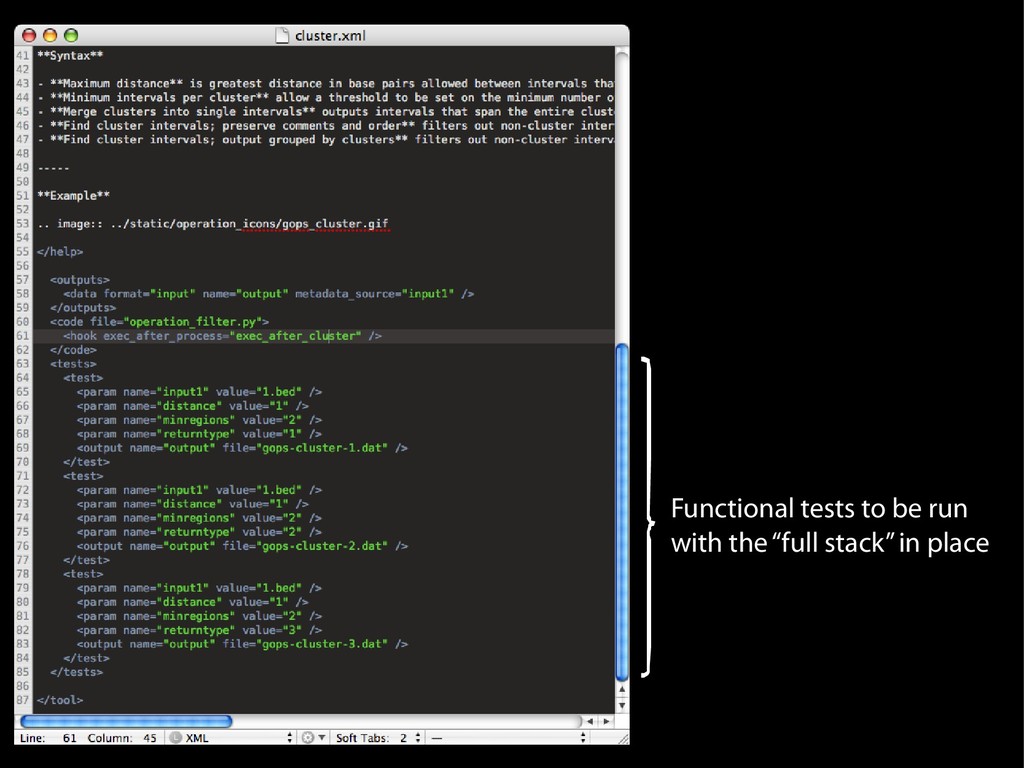{kind=link}
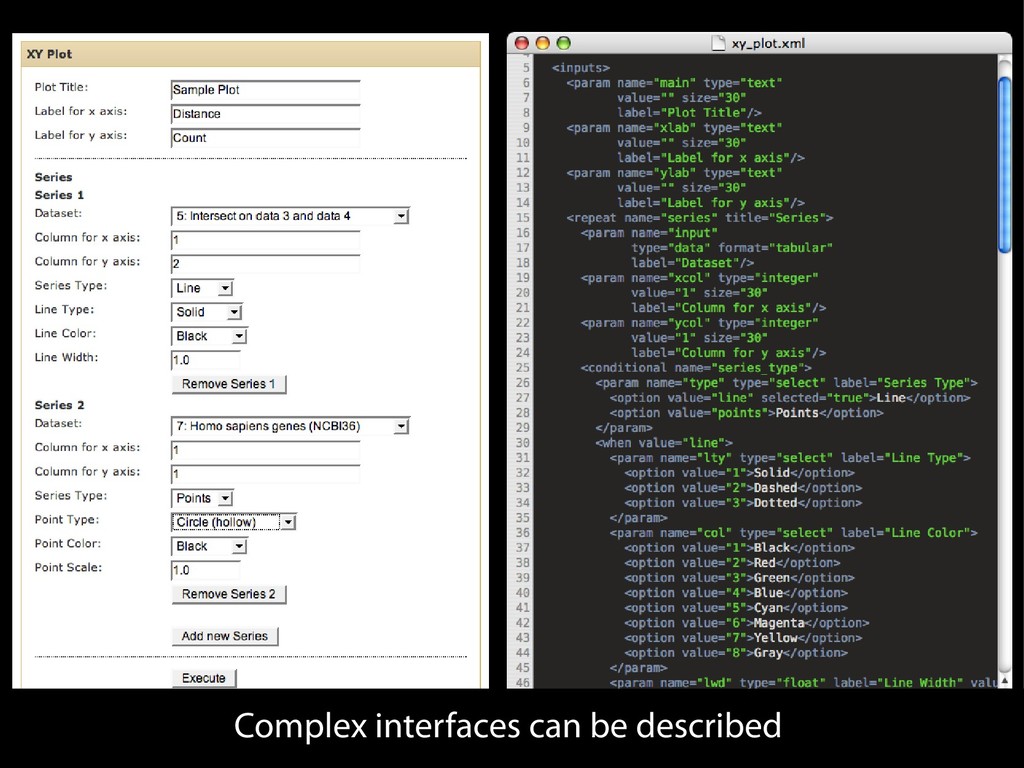{kind=link}
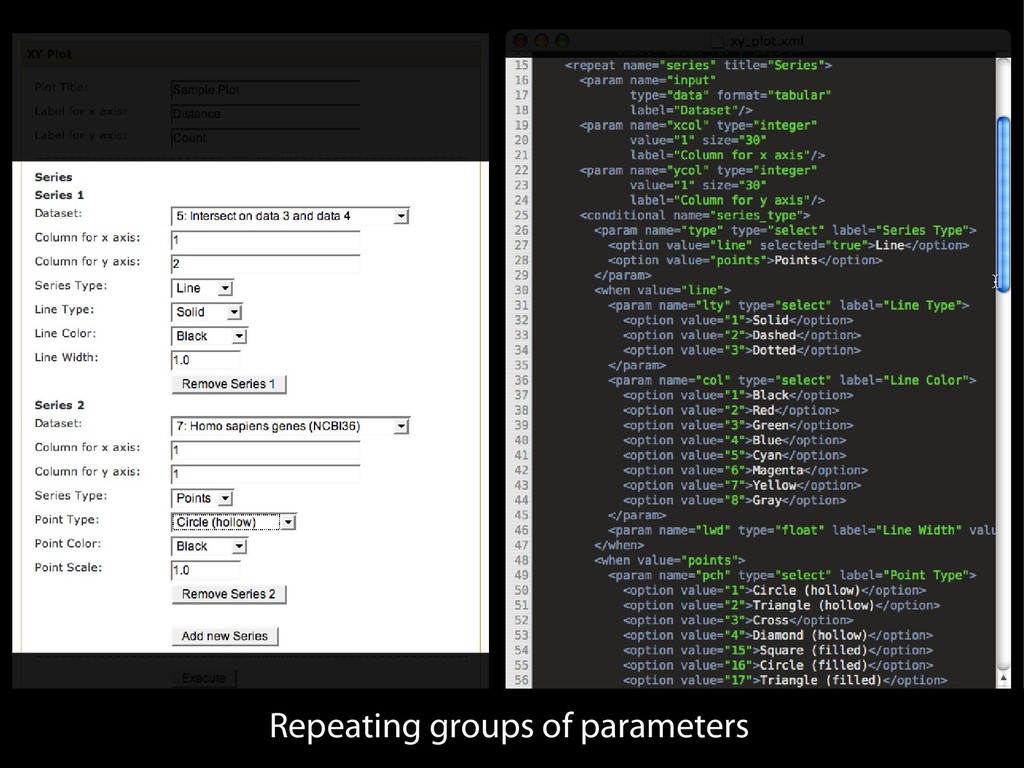{kind=link}
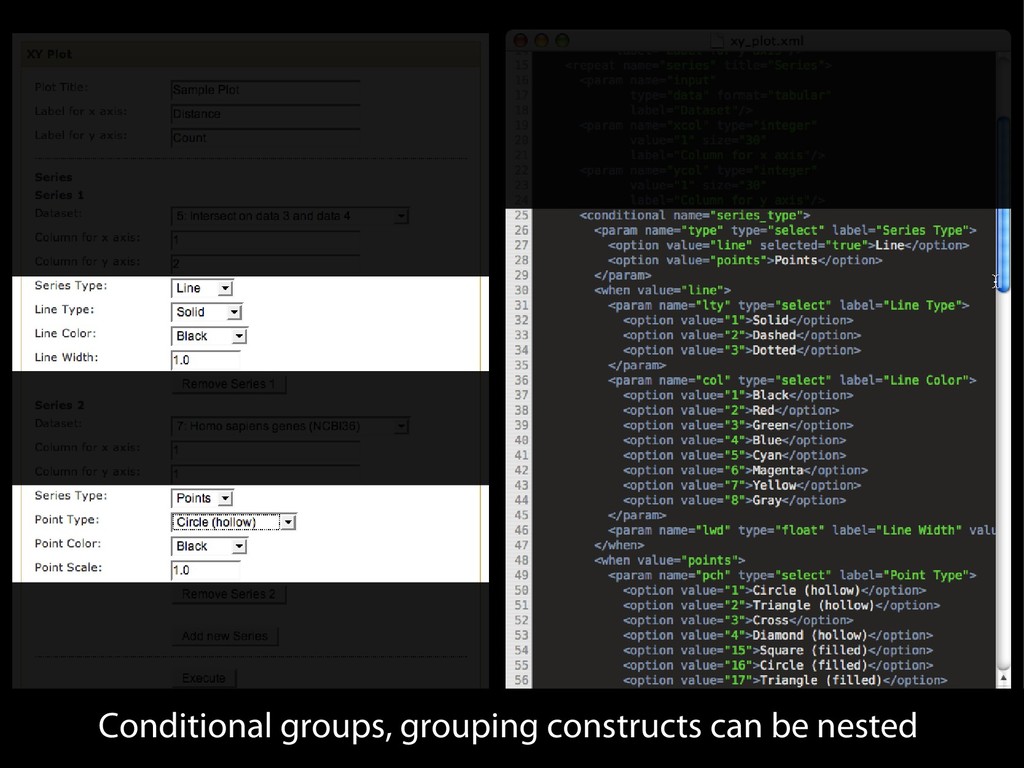{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
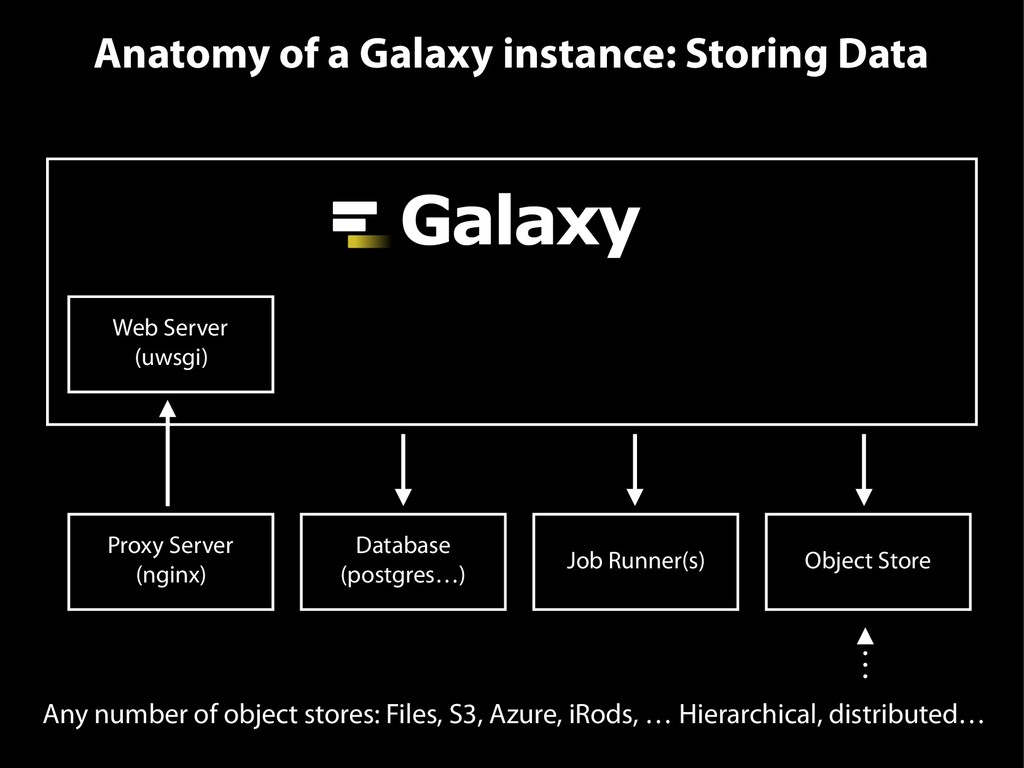{kind=link}
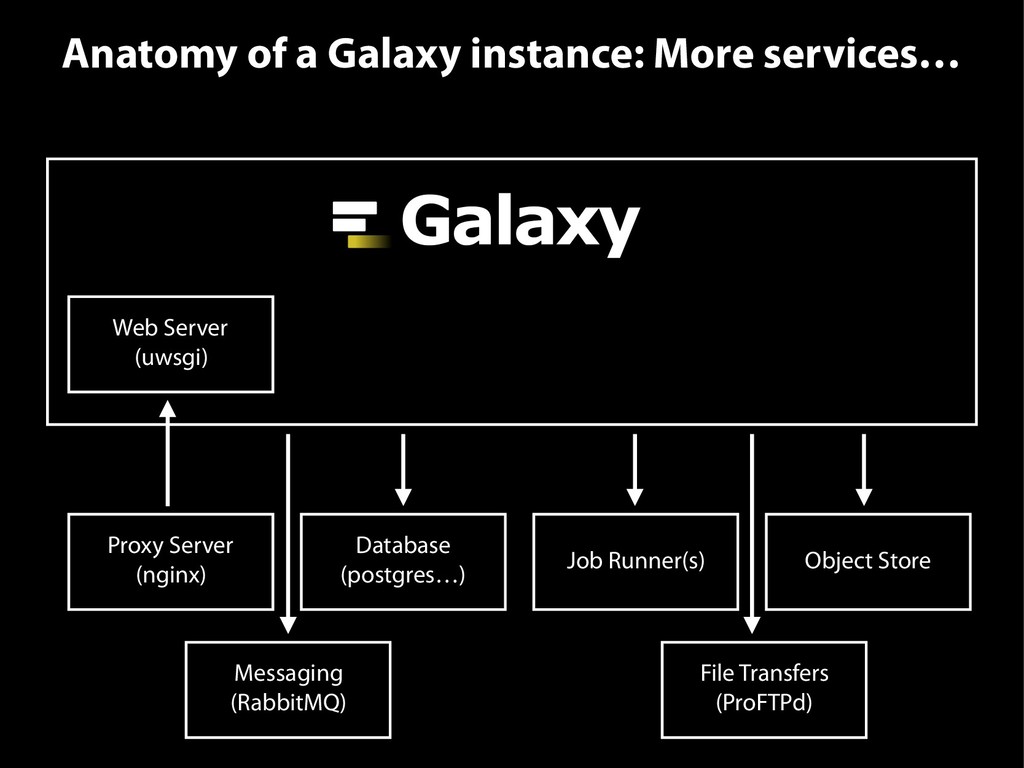{kind=link}
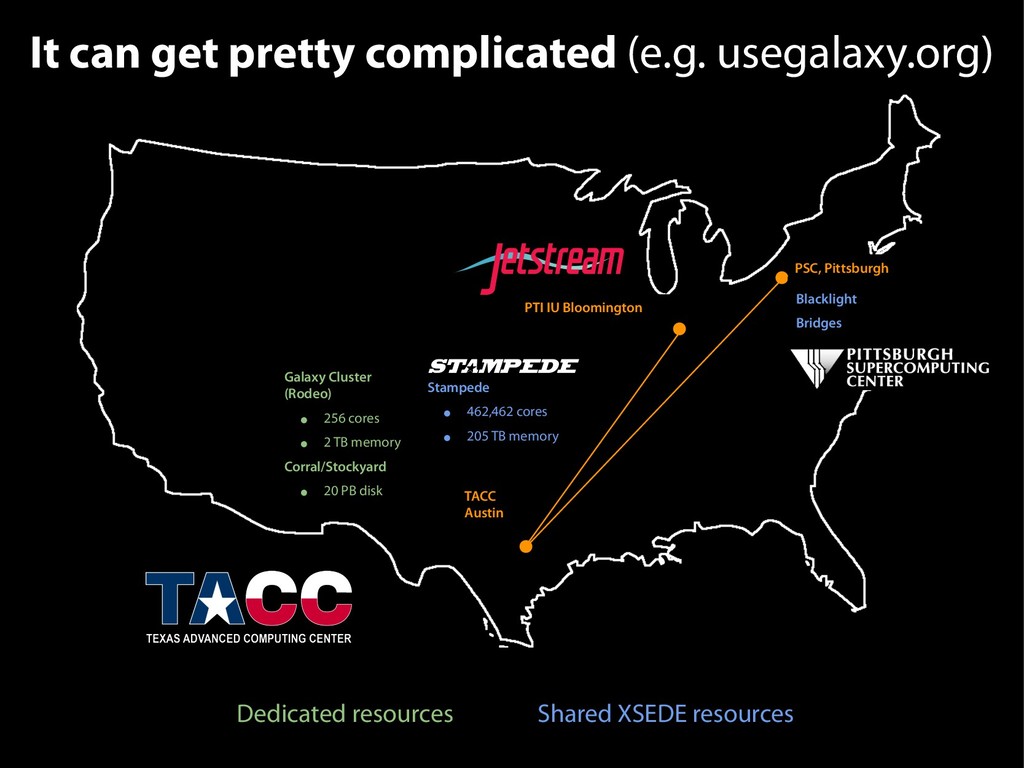{kind=link}
{kind=link}
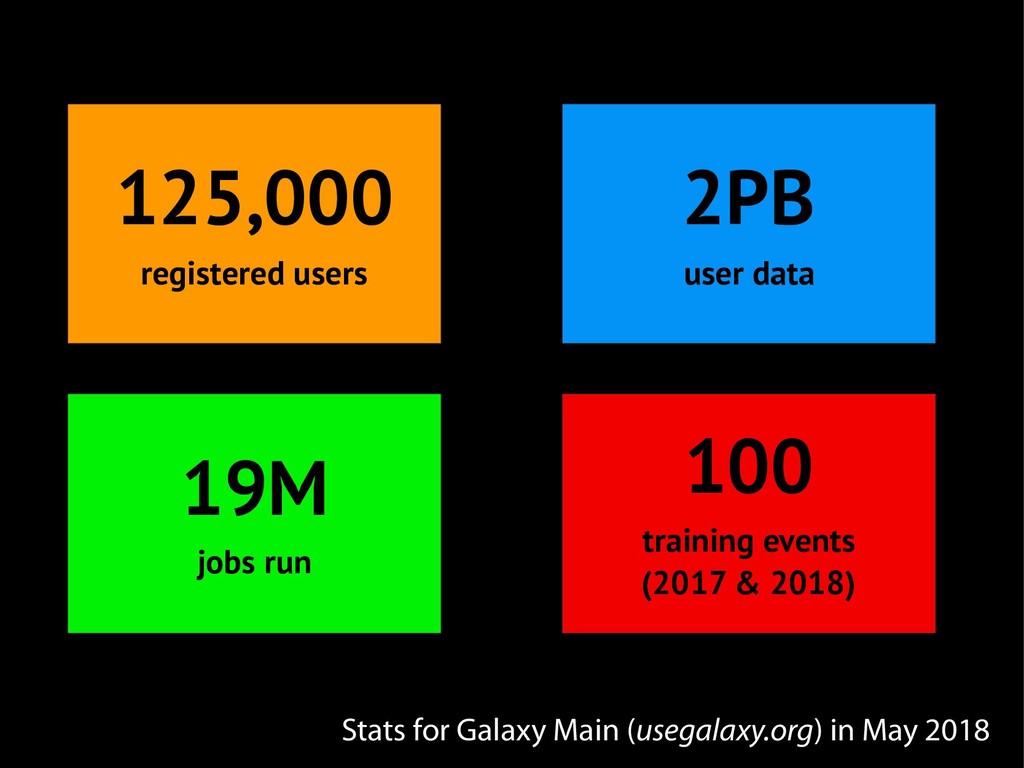{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}