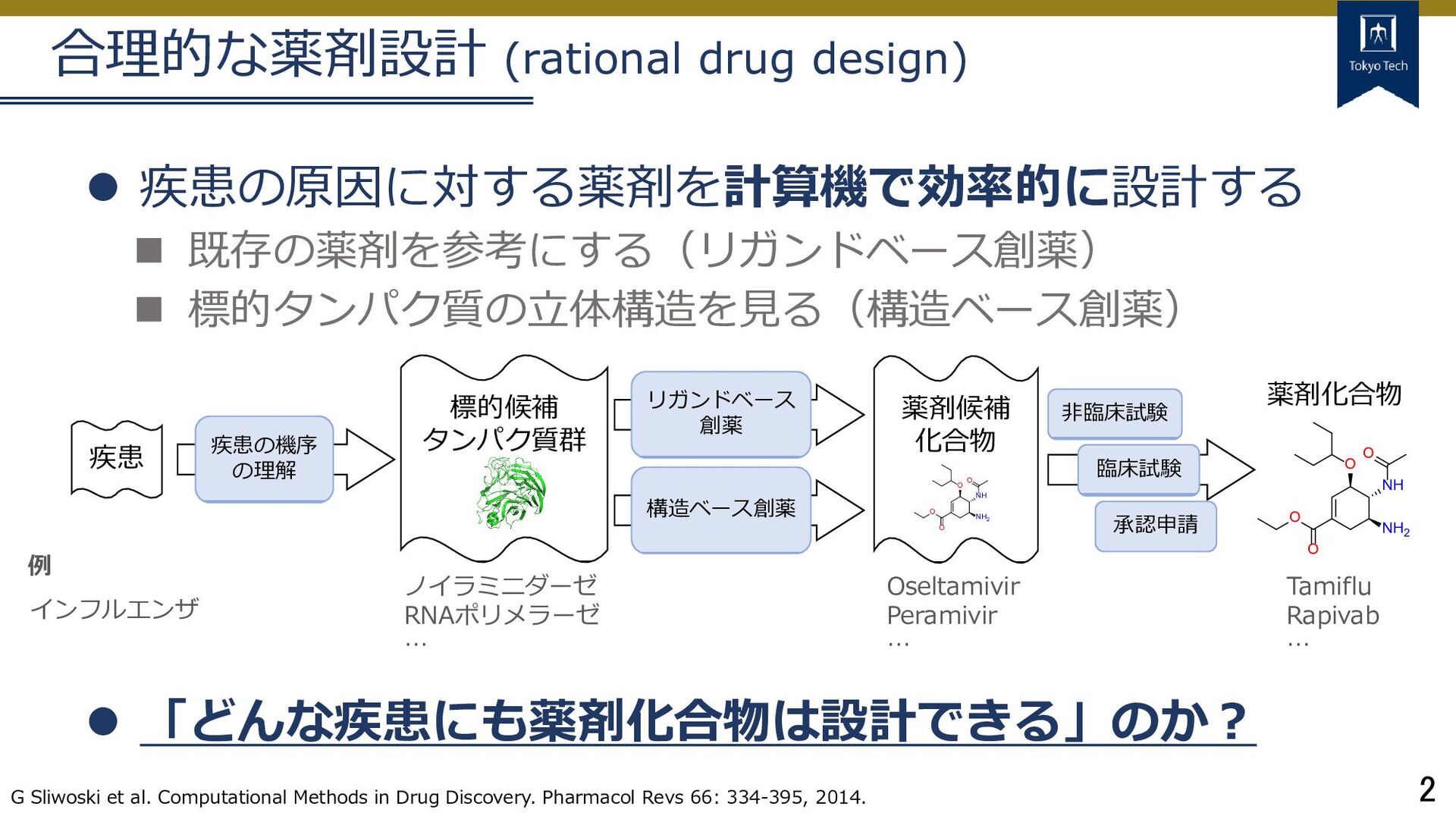
標的タンパク質の立体構造を見る(構造ベース創薬) 「どんな疾患にも薬剤化合物は設計できる」のか? G Sliwoski et al. Computational Methods in Drug Discovery. Pharmacol Revs 66: 334-395, 2014. 疾患 標的候補 タンパク質群 疾患の機序 の理解 リガンドベース 創薬 構造ベース創薬 薬剤候補 化合物 非臨床試験 臨床試験 承認申請 インフルエンザ 例 ノイラミニダーゼ RNAポリメラーゼ … Oseltamivir Peramivir … O NH O NH2 O O O NH O NH2 O O Tamiflu Rapivab … 薬剤化合物
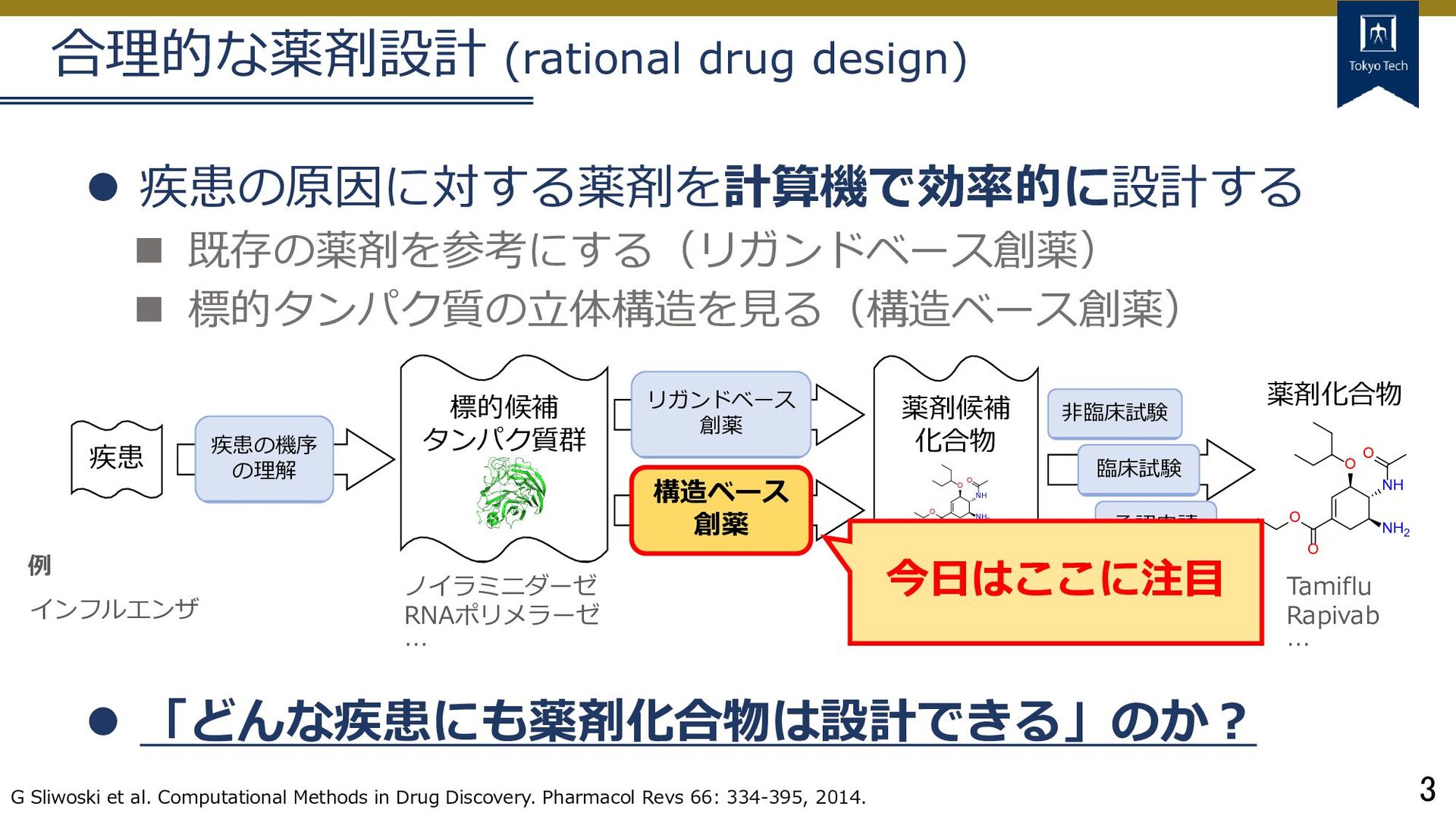
標的タンパク質の立体構造を見る(構造ベース創薬) 「どんな疾患にも薬剤化合物は設計できる」のか? G Sliwoski et al. Computational Methods in Drug Discovery. Pharmacol Revs 66: 334-395, 2014. 疾患 標的候補 タンパク質群 疾患の機序 の理解 リガンドベース 創薬 構造ベース 創薬 薬剤候補 化合物 非臨床試験 臨床試験 承認申請 インフルエンザ 例 ノイラミニダーゼ RNAポリメラーゼ … Oseltamivir Peramivir … O NH O NH2 O O O NH O NH2 O O Tamiflu Rapivab … 薬剤化合物 今日はここに注目
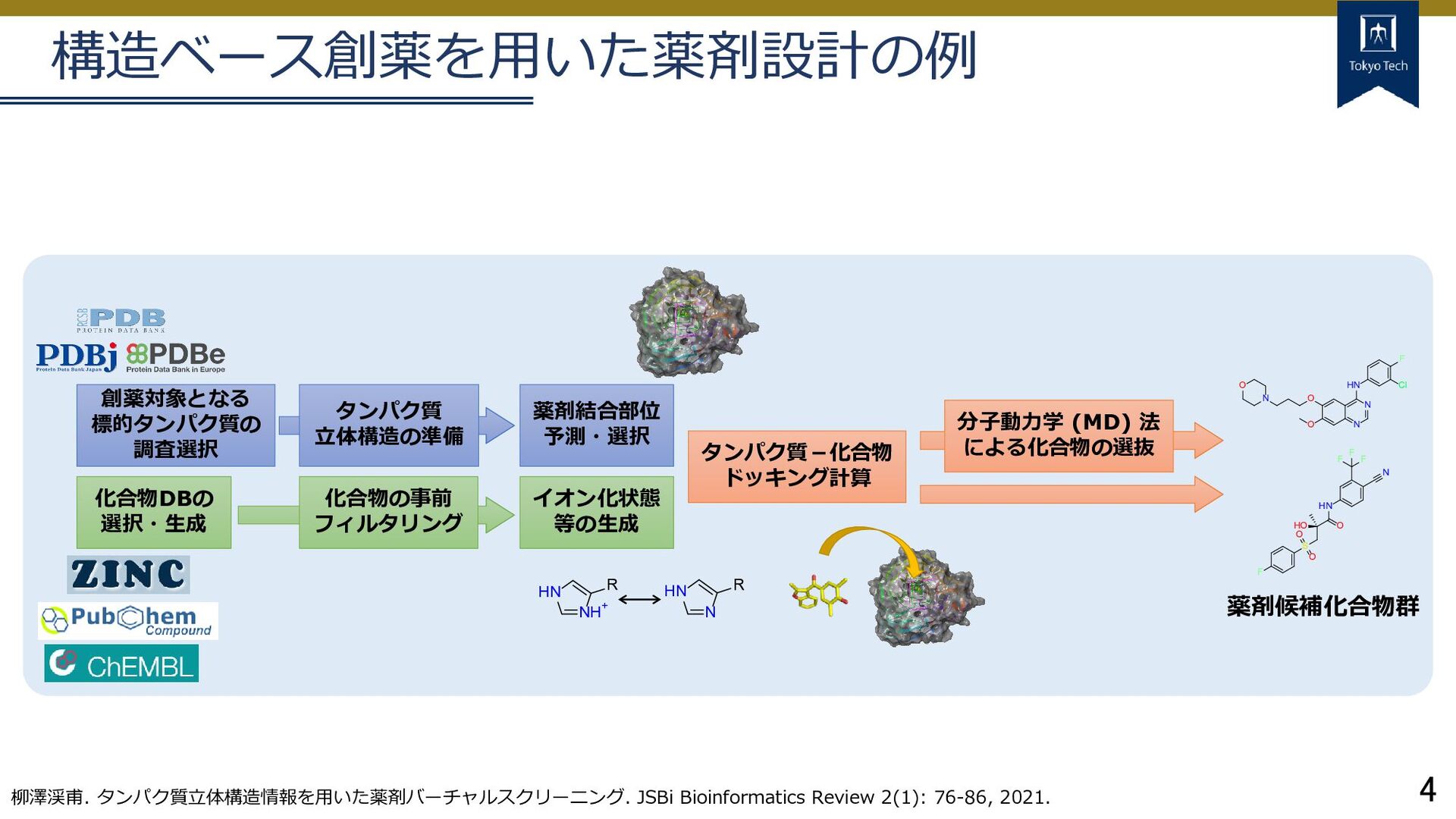
創薬対象となる 標的タンパク質の 調査選択 薬剤結合部位 予測・選択 タンパク質-化合物 ドッキング計算 化合物の事前 フィルタリング イオン化状態 等の生成 化合物DBの 選択・生成 分子動力学 (MD) 法 による化合物の選抜 O N N HN F Cl O N O HO S O O F O HN N F F F 薬剤候補化合物群 NH+ HN R HN N R タンパク質 立体構造の準備
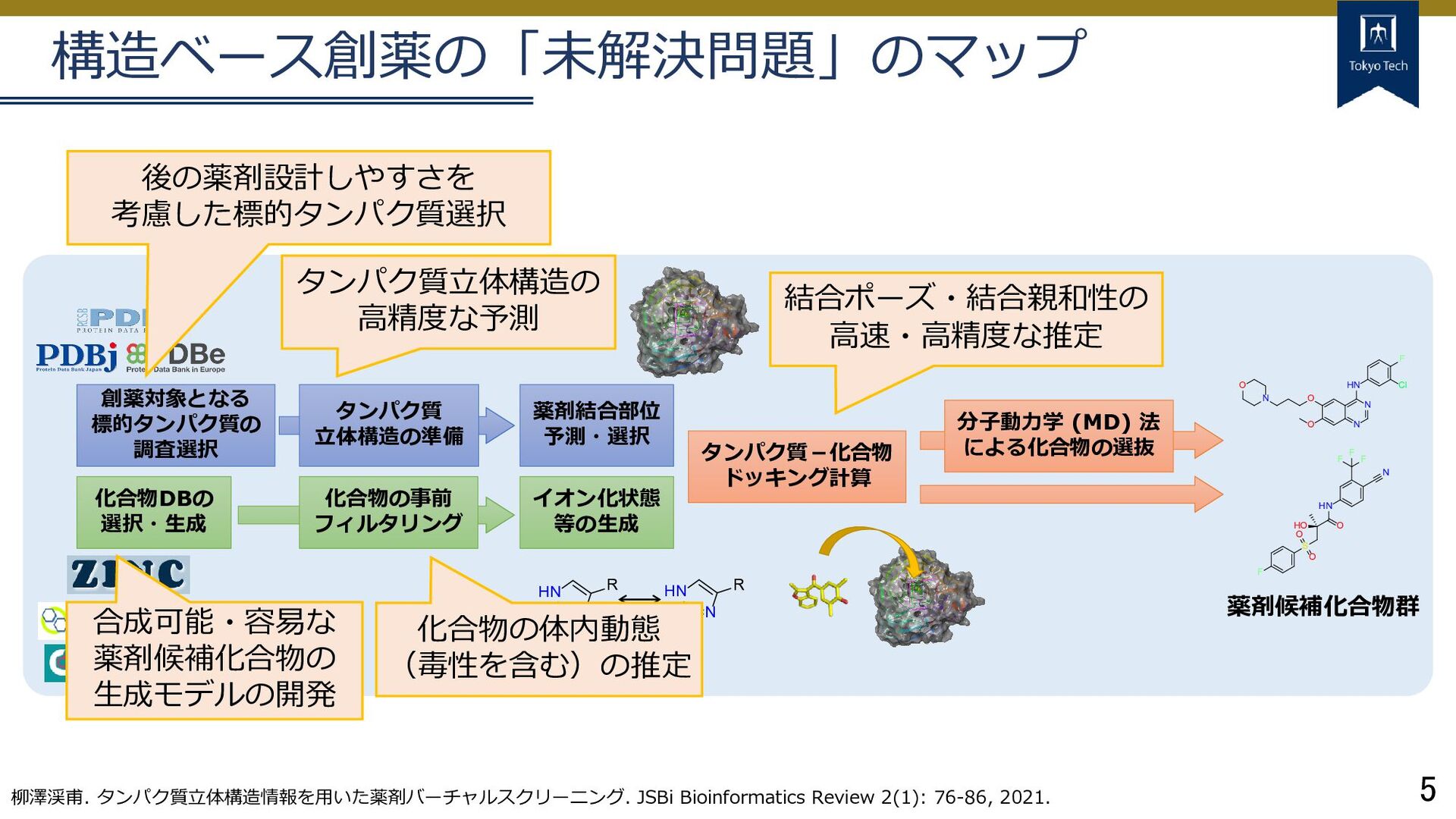
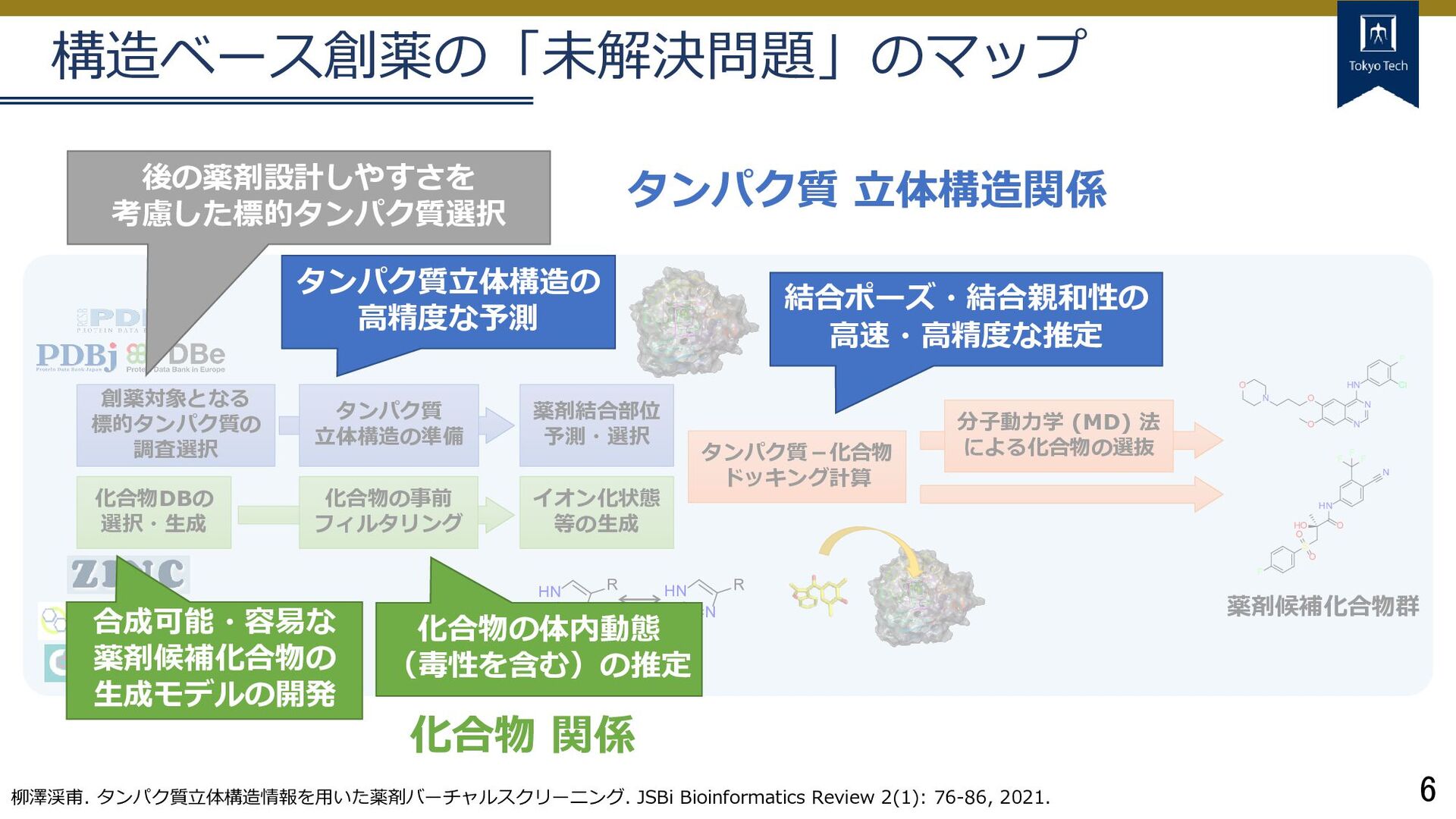
イオン化状態 等の生成 化合物DBの 選択・生成 分子動力学 (MD) 法 による化合物の選抜 O N N HN F Cl O N O HO S O O F O HN N F F F 薬剤候補化合物群 NH+ HN R HN N R タンパク質 立体構造の準備 構造ベース創薬の「未解決問題」のマップ 柳澤渓甫. タンパク質立体構造情報を用いた薬剤バーチャルスクリーニング. JSBi Bioinformatics Review 2(1): 76-86, 2021. タンパク質立体構造の 高精度な予測 結合ポーズ・結合親和性の 高速・高精度な推定 後の薬剤設計しやすさを 考慮した標的タンパク質選択 合成可能・容易な 薬剤候補化合物の 生成モデルの開発 化合物の体内動態 (毒性を含む)の推定
イオン化状態 等の生成 化合物DBの 選択・生成 分子動力学 (MD) 法 による化合物の選抜 O N N HN F Cl O N O HO S O O F O HN N F F F 薬剤候補化合物群 NH+ HN R HN N R タンパク質 立体構造の準備 構造ベース創薬の「未解決問題」のマップ 柳澤渓甫. タンパク質立体構造情報を用いた薬剤バーチャルスクリーニング. JSBi Bioinformatics Review 2(1): 76-86, 2021. タンパク質立体構造の 高精度な予測 結合ポーズ・結合親和性の 高速・高精度な推定 後の薬剤設計しやすさを 考慮した標的タンパク質選択 合成可能・容易な 薬剤候補化合物の 生成モデルの開発 化合物の体内動態 (毒性を含む)の推定 タンパク質 立体構造関係 化合物 関係
成功率 𝒑𝒑 < 𝟎𝟎. 𝟎𝟎𝟎𝟎𝟎𝟎 𝒑𝒑 > 𝟎𝟎. 𝟎𝟎𝟎𝟎 青:実験的に得られた holo 立体構造を利用 橙:AlphaFoldで生成された構造を利用 緑:従来法(template-base)で 生成された構造を利用 論文[1]より抜粋、一部加筆修正 [1] M Karelina et al. How accurately can one predict drug binding modes using AlphaFold models? eLife, 2023. (epub ahead of print)
AlphaFold × simulation で互いに補うのが今は現実的? ドッキングと分子動力学法を繰り返し行うことで そのまま利用するより精度向上することが報告されている [2] [1] M L Hekkelman et al. AlphaFill: enriching AlphaFold models with ligands and cofactors. Nat Methods 20: 205-213, 2022. [2] Y Zhang et al. Benchmarking Refined and Unrefined AlphaFold2 Structures for Hit Discovery. J Chem Inf Model 63(6): 1656-1667, 2023. 論文[1]より抜粋、一部加筆修正 AlphaFold AlphaFill
[2] ことで 「生体内における化合物の類似性」を見れるはず 様々な計算機実験を通して より効率的な化合物表現を 模索することも重要 現在は文字列表現が主流 [1] H Iwata et al. Predicting Total Drug Clearance and Volumes of Distribution Using the Machine Learning-Mediated Multimodal Method through the Imputation of Various Nonclinical Data. J Chem Inf Model 62(17): 4057-4065, 2022. [2] N Moshkov et al. Predicting compound activity from phenotypic profiles and chemical structures. Nat Commun 14:1967, 2023. 論文[2]より抜粋
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![8 AlphaFoldの現状 AlphaFoldはタンパク質単品の立体構造を高精度に予測 しかし、この予測構造は ドッキング計算の精度向上には 寄与しない との報告あり [1] 化合物との複合体構造を直接予測しないと効果は限定的 ドッキング](https://files.speakerdeck.com/presentations/1c7af783a3da49b7a3da13c450a06150/slide_7.jpg){kind=link}
![9 AlphaFoldを複合体構造予測に拡げていく 複合体構造(&結合親和性)を予測できる手法が望まれる (当然)様々な研究グループが挑戦しているが、 既知複合体構造の少なさにより困難 補因子やイオンなどに限れば、 用いる配列・テンプレート構造を工夫可能 [1]](https://files.speakerdeck.com/presentations/1c7af783a3da49b7a3da13c450a06150/slide_8.jpg){kind=link}
![10 より良い化合物の潜在空間を構築する 化合物に対する実験データの量には限りがある[1] 実験コストが高い & お金になるので門外不出になりやすい マルチモーダルでより良い化合物潜在空間を見つけ出す? オミックス解析などと組み合わせる](https://files.speakerdeck.com/presentations/1c7af783a3da49b7a3da13c450a06150/slide_9.jpg){kind=link}
{kind=link}