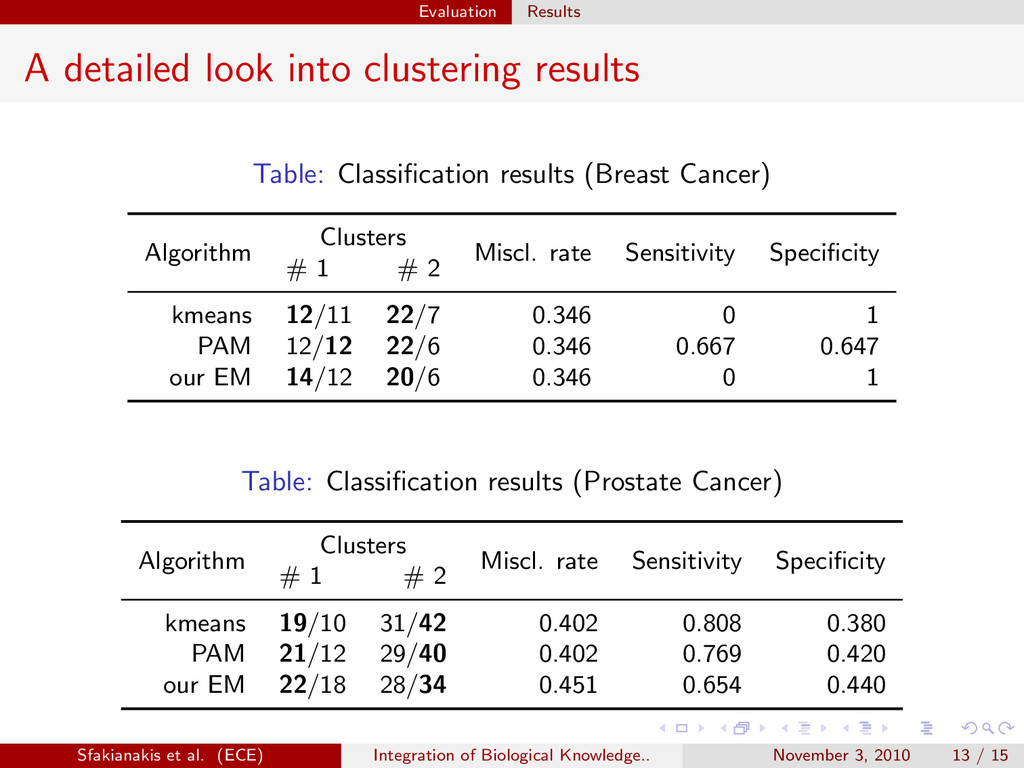
sparse structure of the cov. matrix is imposed on every component of the mixture model so that component densities are rewritten as fi (x x xj ;θi θi θi ) = N(x x xj ;µ µ µi ,Σ Σ Σi ) (7) and then taking into account the block diagonal structure we get a factorization of the form: fi (x x xj ;θi θi θi ) = N(x x x(r) j ;µ µ µ(r) i ,D D D(r) i ) K ∏ k=1 N(x x x(k) j ;µ µ µ(k) i ,Σ Σ Σ(k) i ) (8) and the mixture density becomes: f (x x xj ;Θ Θ Θ) = g ∑ i=1 πi N(x x x(r) j ;µ µ µ(r) i ,D D D(r) i ) · K ∏ k=1 N(x x x(k) j ;µ µ µ(k) i ,Σ Σ Σ(k) i ) = g ∑ i=1 πi K+1 ∏ k=1 N(x x x(k) j ;µ µ µ(k) i ,Σ Σ Σ(k) i ) (9) Sfakianakis et al. (ECE) Integration of Biological Knowledge.. November 3, 2010 7 / 15
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}