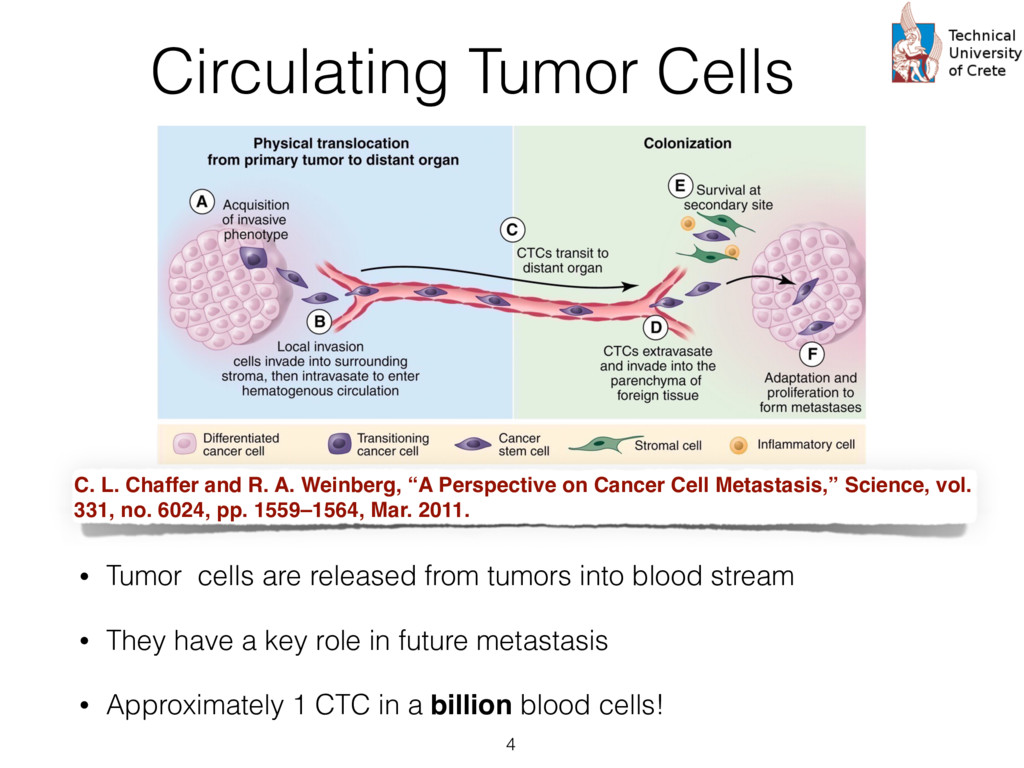
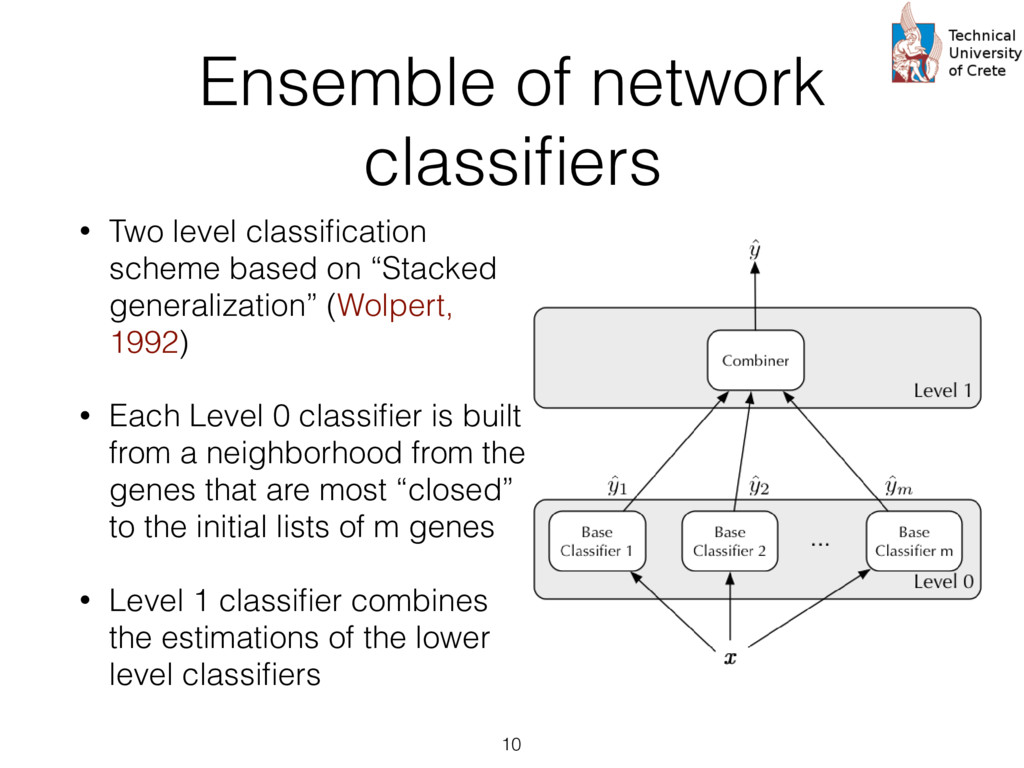
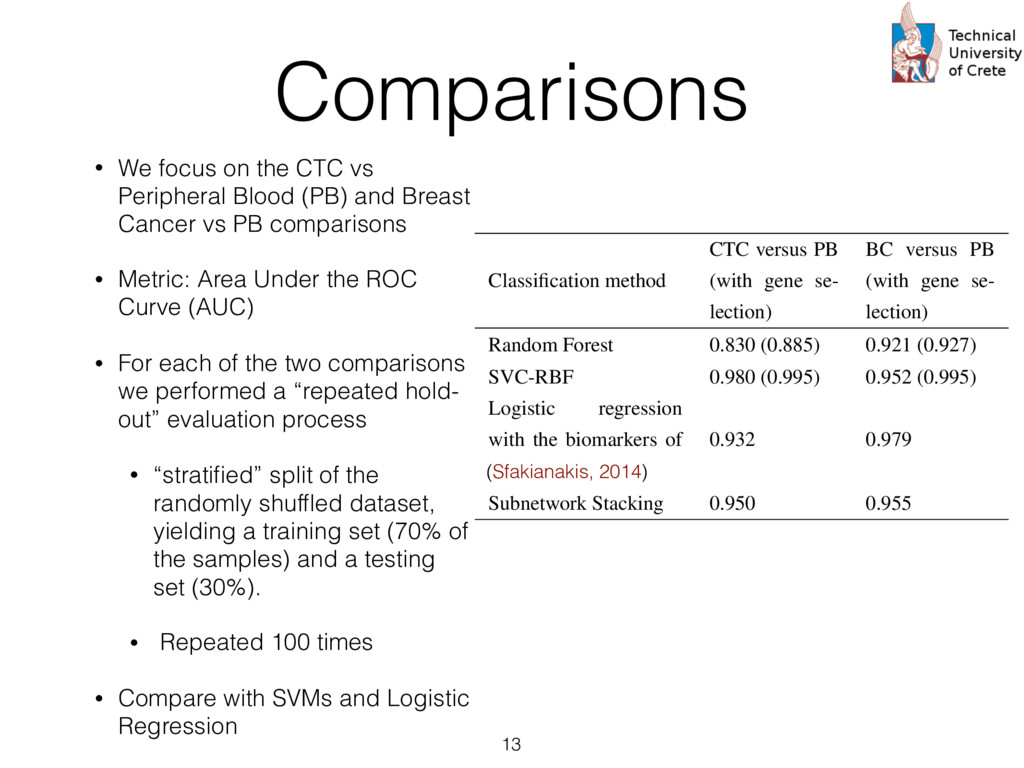
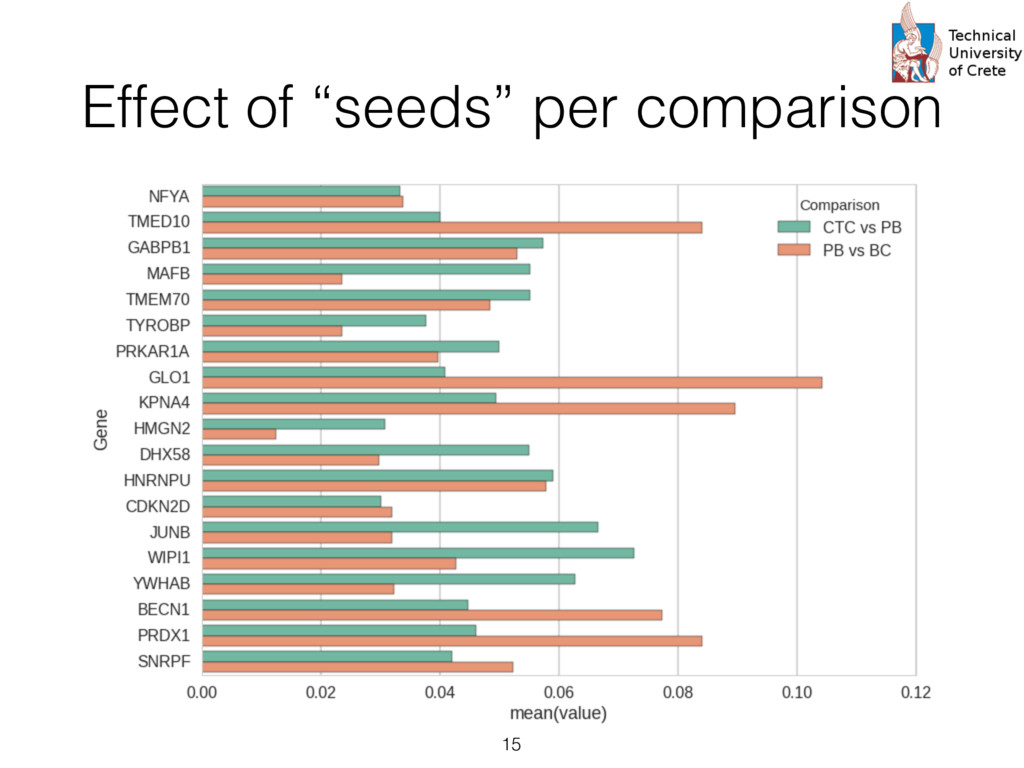
expression profiles from peripheral blood, circulating cancer, breast cancer tissue, and normal epithelia • Probes are summarized according to UniGene clusters : Stacked generalization or 2-level “stacking” of classifiers. The s at the first level (Level 0) take as input the input cases and each them produces a prediction. The predictions of the first level rs are then given as input to the second level (Level 1) classifier (“combiner”) that provides the final prediction. s trained using Logistic Regression. The rationale his choice is twofold: First, logistic regression pro- s a probabilistic output, i.e. a level of confidence that nput sample belongs to the “positive” class (e.g. a or prognosis” group) that can be used as a numeri- eature for the second level classifier. Secondly, the abilistic output of a logistic regression classifier is lly well calibrated since it optimizes directly the log [18]. The choice of how many neighbors to consider each gi , which equivalently means the radius of the en ball” [19] centered at gi , is determined by an in- al cross validation. In this cross validation, increasing es of the radius r , that is the distance from the center e gi , are used and the subset of genes contained in said distance are checked in terms of the impact on sification performance as measured by the Matthews elation coefficient [20]. The different values of r tried A. Data We have used the recently available data set of Lang e al [21] to test the proposed two-level classification approac This data set is available as three “sub-series” in the Gen Expression Omnibus (GEO) public database as the “supe series” GSE45965 2. It consists of 67 gene expression pro files from peripheral blood, circulating cancer, breast cance tissue, and normal epithelia, as shown in Table 1. Table 1: Number of sample and characteristics of the public dataset GSE45965 Phenotype Number of samples Normal Peripheral Blood 8 Breast Cancer Tumor 50 Circulating Tumor Cells 5 Normal Epithelia 4 For the proper annotation of the gene probes in the data s we have used the UniGene database3 to perform mapping from the GeneBank identifiers to the Entrez Gene ids an gene symbols. Probes that dont’t have a UniGene annotatio or that have not been measured in all samples were remove For the case where multiple probes map to the same Entre Gene identifier the average (mean) expression value was ca culated and used in the downstream analysis. After this an notations and summarization step, we are left with aroun 15,000 unique genes. http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE45965 cate” instead of just considering their immediate neigh- We then consider a set of input genes that are used as for building a corresponding set of “base classifiers” d on the direct or indirect interactions the seeds have other genes. The base classifiers are subsequently used ain a second level classifier that can potentially com- ntelligently the successes and failures of them. The im- entation is based on the “scikit-learn” machine learn- ramework for Python [27] and all the code and the can be found at https://github.com/sgsfak/ net_stacking . e use of biological networks and random walks in hs have been studied in multiple publications [28, 29, 1]. Especially HotNet2 [29] uses similar random walk- d schemes, but our aim is to construct an adaptive, gene ture initialized, and biologically driven classifier. The s are indeed promising and make stronger the selection r initial gene list used for the initialization, but further ation, tuning, and validation are needed. ACKNOWLEDGMENTS is research is partially supported by the “ONCOSEED” ct funded by the NSRF2007-13 Program of the Greek stry of Education, Lifelong Learning and Religious Af- CONFLICT OF INTEREST Reviews Cancer. 2007;7:545–553. 9. Chung F. The heat kernel as the pagerank of a graph Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19735–19740. 10. Can T, C ¸ amolu O, Singh A K. Analysis of protein-protein interaction networks using random walks in Proceedings of the 5th international workshop on Bioinformatics:61–68ACM 2005. 11. Lovasz L. Random walks on graphs: A survey Combinatorics. 1993. 12. Kittler J, Hatef M, Duin R P W, Matas J. On Combining Classifiers. IEEE Trans. Pattern Anal. Mach. Intell. (). 1998;20:226–239. 13. Dietterich T G. Ensemble Methods in Machine Learning. Multiple Classifier Systems. 2000:1–15. 14. Schapire R E. The strength of weak learnability Machine learning. 1990;5:197–227. 15. Freund Y, Schapire R E. A decision-theoretic generalization of on-line learning and an application to boosting Journal of computer and system sciences. 1997;55:119–139. 16. Breiman L. Bagging predictors Machine learning. 1996. 17. Wolpert D H. Stacked generalization Neural Networks. 1992;5:241– 259. 18. Bishop C. Pattern recognition and machine learning. New York: Springer 2006. 19. Rosenlicht M. Introduction to analysis. New York: Dover 1986. 20. Powers D M. Evaluation: from precision, recall and F-measure to ROC, informedness, markedness and correlation Journal of Machine Learn- ing Technologies. 2011;2:37–63. 21. Lang J E, Scott J H, Wolf D M, et al. Expression profiling of circulat- ing tumor cells in metastatic breast cancer Breast cancer research and treatment. 2015;149:121–131. 22. Fawcett T. An introduction to ROC analysis Pattern Recognition Let- ters. 2006;27:861–874. 23. Breiman L. Random Forests Machine Learning. 2001;45:5–32. 24. Subramanian A, Tamayo P, Mootha V K, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. 25. Mosca E, Alfieri R, Merelli I, Viti F, Calabria A, Milanesi L. A mul- tilevel data integration resource for breast cancer study BMC Systems Biology. 2010;4:76. 26. Chuang H-Y, Lee E, Liu Y-T, Lee D, Ideker T. Network-based classifi- 12
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}