sequence annotation. • Understand the concept of chromatin states as combinatorial patterns of regulatory signals. • Learn how HMMs can be applied to annotate regulatory features of the genome.
directly followed by “G” • CpG sites are targets of DNA methylation • Addition of CH3 in C-nucleotides • Roles in gene regulation (typically silenced) • Mechanism of epigenetic memory • But… CpG sites have a tendency to get mutated • CG à TG • Therefore, “CG” dinucleotides are rare in vertebrate genomes • CpG islands: regions with many CpG sites • >200bp • GC content greater than usual • “CG” dinucleotides more common than rest of genome • Typically near gene promoters • Seem to be protected from DNA methylation
detection ATTAGCTTCTAGCTAGCTCGCGTCGCGCGTACGCGCGCGTGACGCTAGATTATATGAGTACGATGCGAC We need a model to represent the statistical properties of CpG islands, and to distinguish them from the rest of the genome.
dice with 4 sides: A, C, G, T • What is the probability of “rolling” each letter? • Probability of a longer sequence: Letter P(letter) A 0.25 C 0.25 G 0.25 T 0.25 P(“GCG”) = P(“G”) x P(“C”) x P(“G”) = 0.25 x 0.25 x 0.25 = 1/64 = 0.015625
dice with 4 sides: A, C, G, T • Probability of a longer sequence: Letter P(letter) A 0.1 C 0.4 G 0.4 T 0.1 P(“GCG”) = P(“G”) x P(“C”) x P(“G”) = 0.4 x 0.4 x 0.4 = 0.064 Note the nucleotides are still independent. No way to represent preference for CG dinucleotides in CpG islands.
two models: • Which model is more likely to generate this sequence: GCG Letter P(letter) A 0.25 C 0.25 G 0.25 T 0.25 Letter P(letter) A 0.1 C 0.4 G 0.4 T 0.1 Model 1 Model 2 Odds ratio: P("GCG"| M2) P("GCG"| M1) = 0.064 0.015625 = 4.096 >1 favors Model 2 Log-odds ratio: log 2 (P("GCG"| M2))− log 2 (P("GCG"| M1))
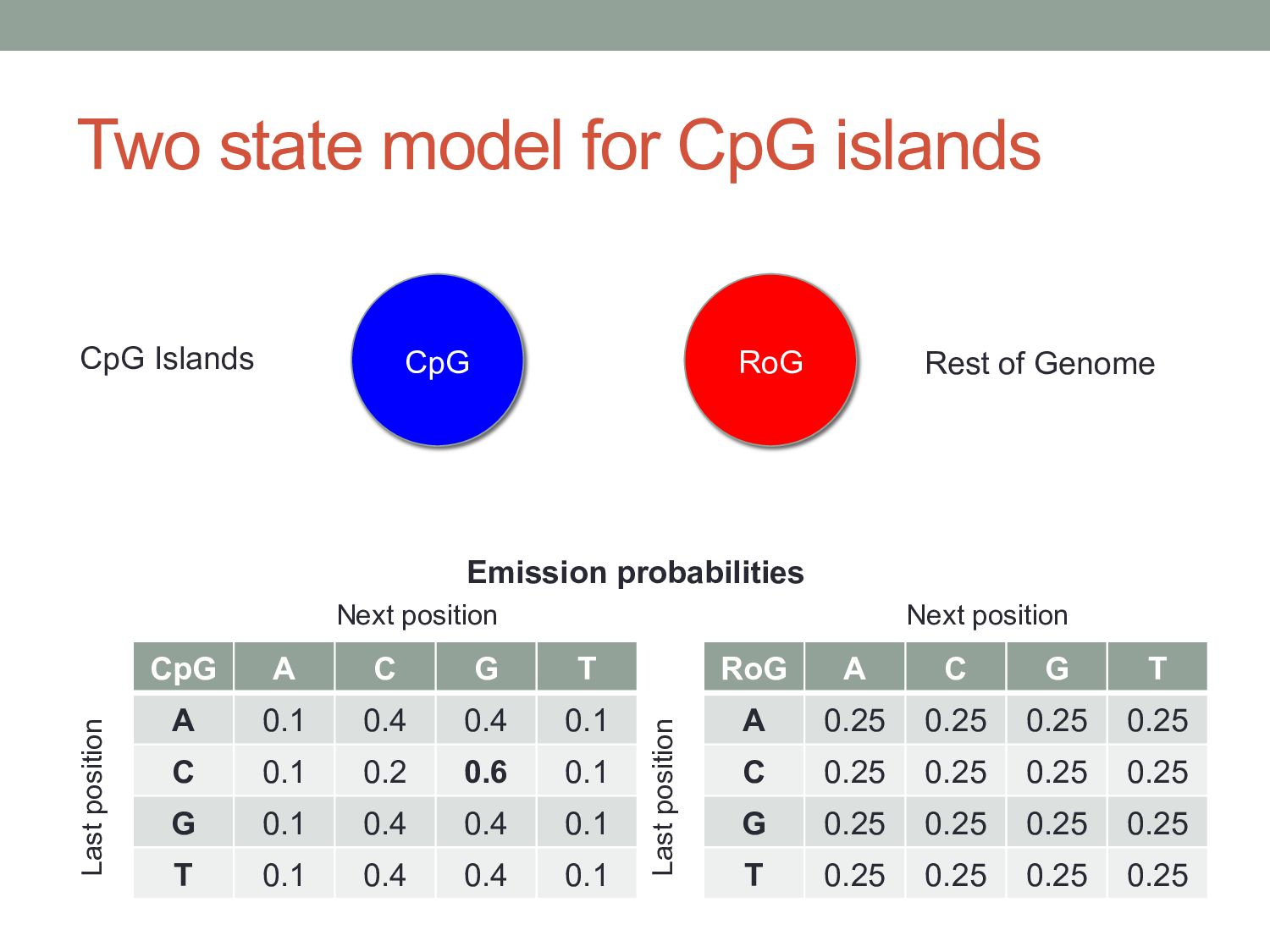
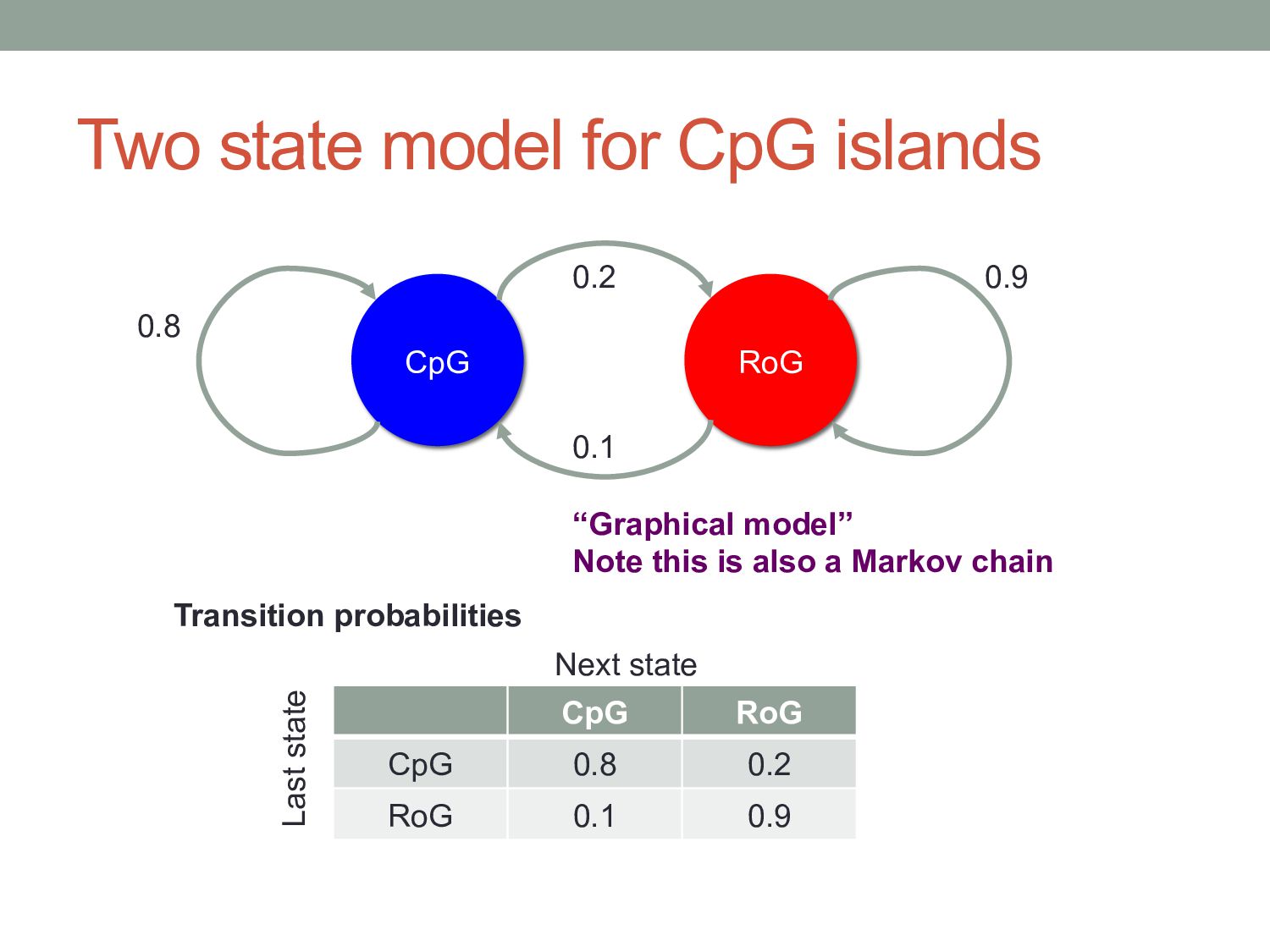
position depends on what you saw at previous position(s). Four possibilities given previous letter = A. Probabilities must sum to 1 A C G T A 0.1 0.4 0.4 0.1 C 0.1 0.2 0.6 0.1 G 0.1 0.4 0.4 0.1 T 0.1 0.4 0.4 0.1 Next position Last position This is a first-order Markov model: the next position depends on the last position. A second-order Markov model would depend on the last two positions, etc.
T A 0.1 0.4 0.4 0.1 C 0.1 0.2 0.6 0.1 G 0.1 0.4 0.4 0.1 T 0.1 0.4 0.4 0.1 Next position Last position RoG A C G T A 0.25 0.25 0.25 0.25 C 0.25 0.25 0.25 0.25 G 0.25 0.25 0.25 0.25 T 0.25 0.25 0.25 0.25 Next position Last position CpG RoG Rest of Genome CpG Islands Emission probabilities
like this are “generative”, because we could use them to “generate” or simulate data with the desired properties. • In the two-state CpG island case, we would • Roll a dice to choose a state (given previous state) • Roll appropriate state’s dice to choose a letter to “emit” (given previous letter) • Example: RoG A G T T C G C G RoG RoG RoG CpG CpG CpG CpG State sequence Emission sequence
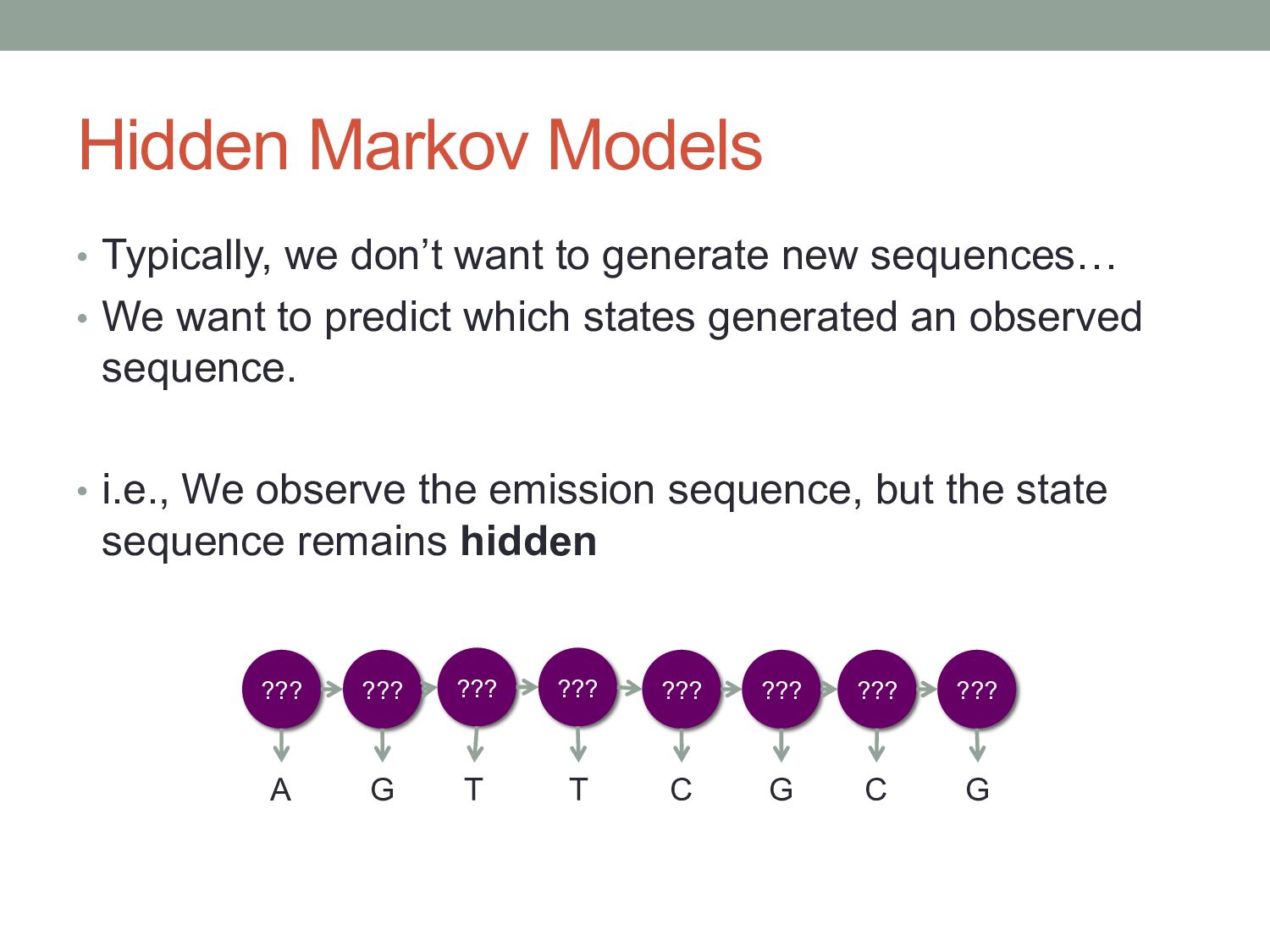
new sequences… • We want to predict which states generated an observed sequence. • i.e., We observe the emission sequence, but the state sequence remains hidden ??? A G T T C G C G ??? ??? ??? ??? ??? ??? ???
feature that occurs in your data. • Hidden Markov Model: predict the state sequence that best explains a sequence of observed data. • Examples: • CpG island annotation • Gene-finding & splice sites • Promoter-finding & regulatory sites • Protein domains • Multiple alignments
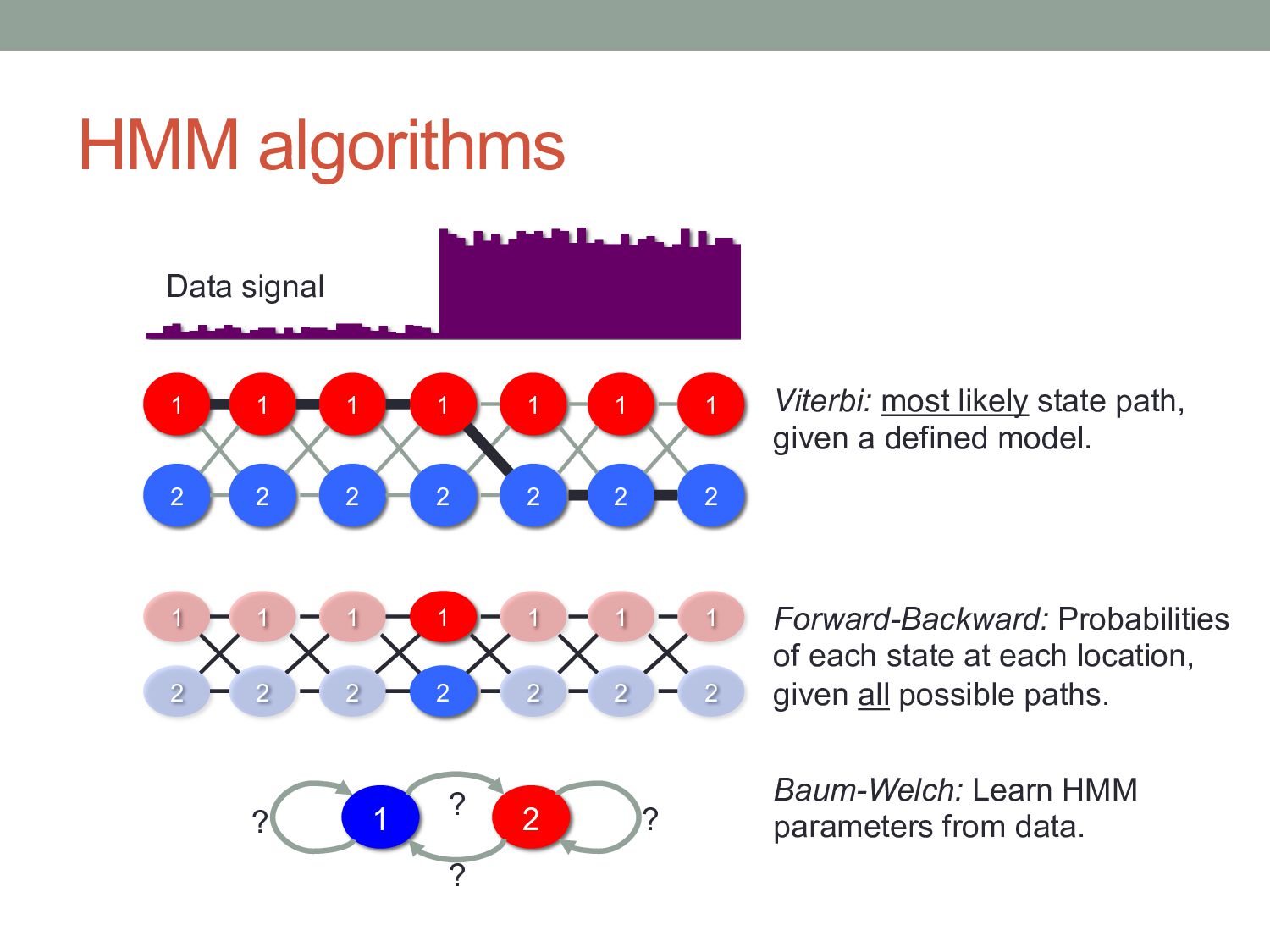
1 2 1 2 1 2 1 1 2 2 1 2 1 2 1 2 1 2 1 2 Viterbi: most likely state path, given a defined model. Forward-Backward: Probabilities of each state at each location, given all possible paths. 1 2 ? ? ? ? Baum-Welch: Learn HMM parameters from data. Data signal
we know all parameters of our Hidden Markov Model (all emission probabilities e, and all transition probabilities a), how could we decode the most likely state path? • Calculate probabilities of all possible state paths? • …won’t that take a while for longer sequences? • Dynamic programming to the rescue! • Recursively calculate the most probable paths ending in each state for each observation i
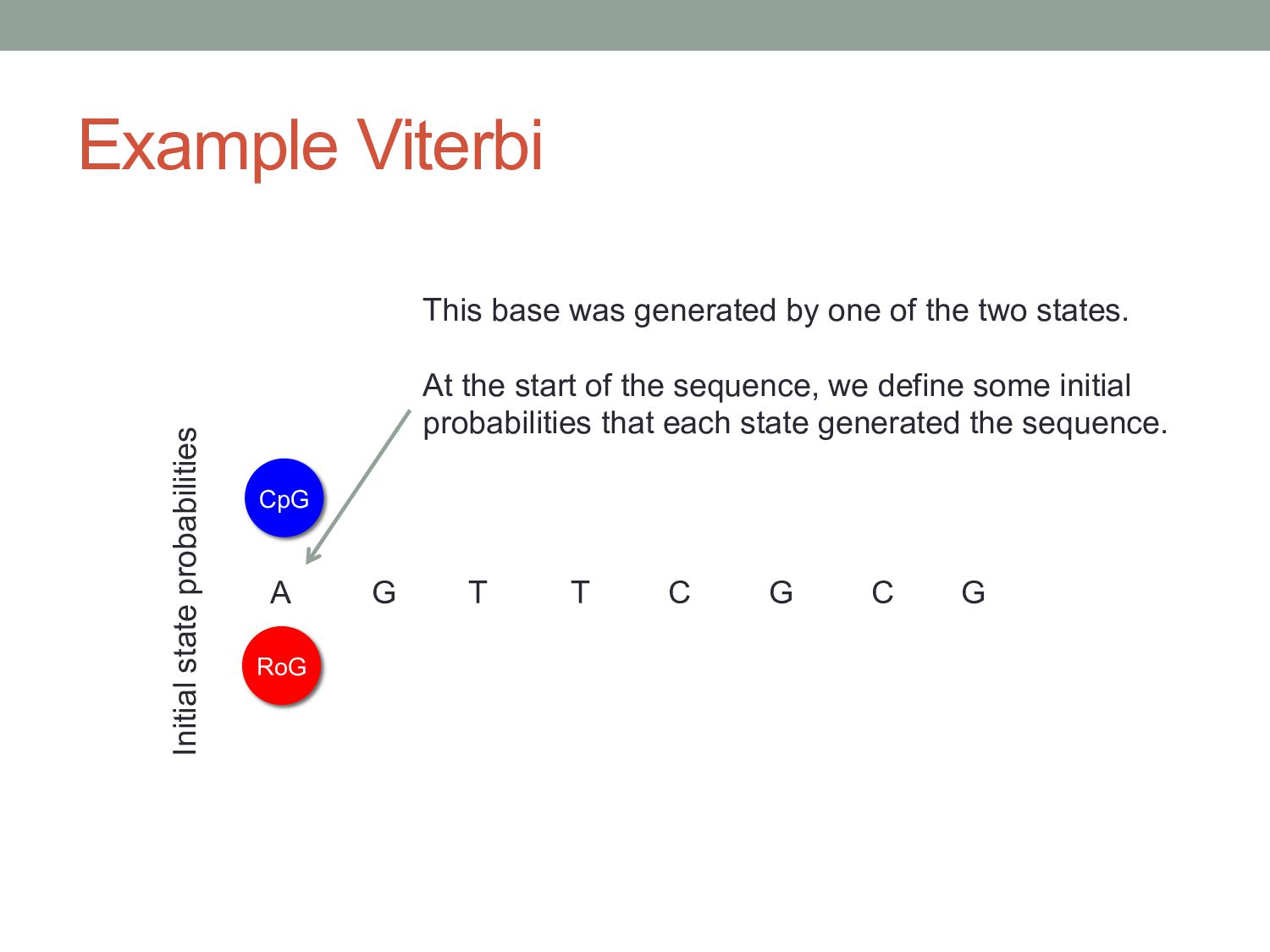
G CpG Initial state probabilities This base was generated by one of the two states. At the start of the sequence, we define some initial probabilities that each state generated the sequence.
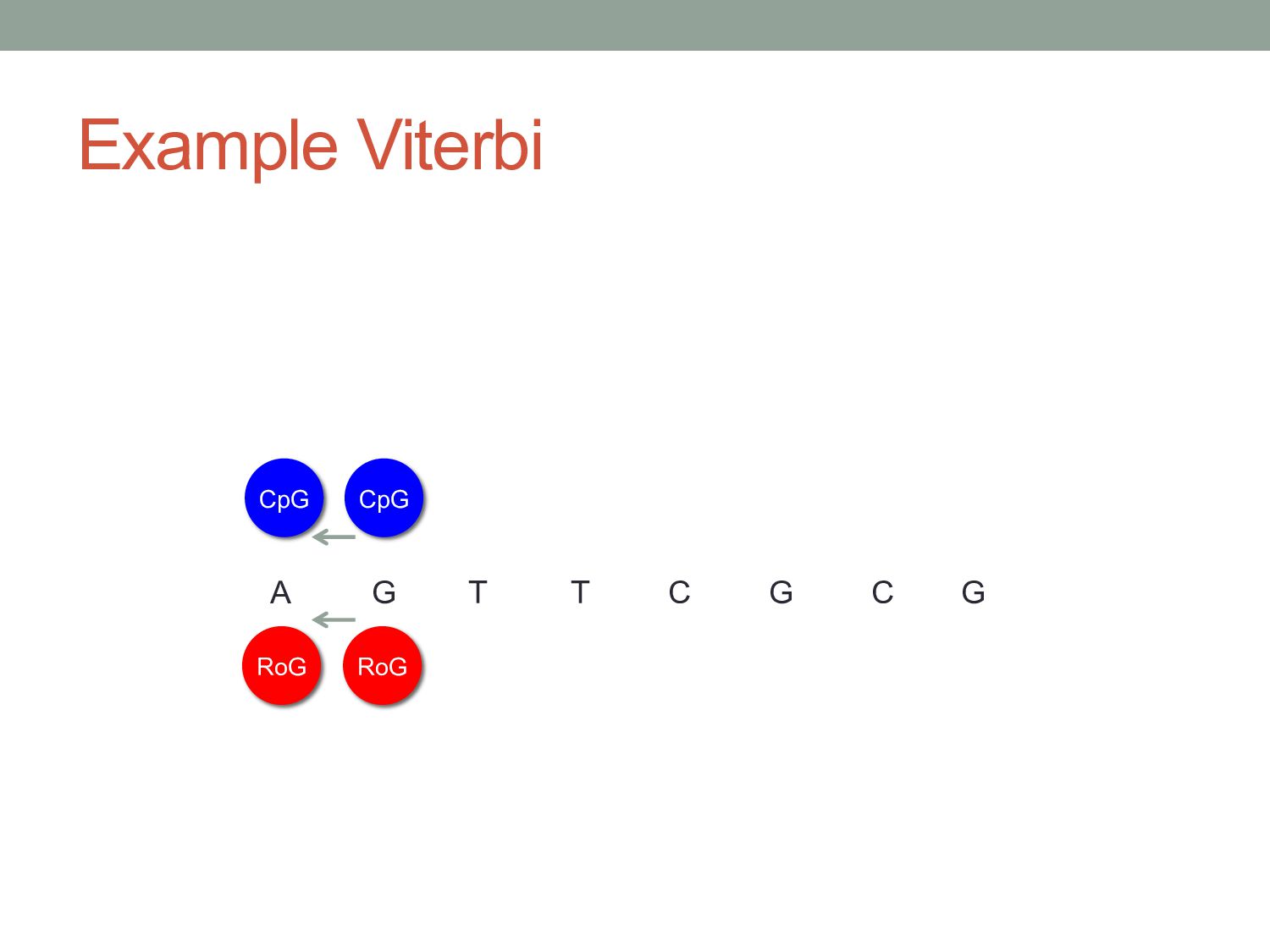
G RoG CpG CpG This base was also generated by one of the two states. Given that we’ve calculated the probabilities of each state generating the previous base, there are only four state transitions that could have led to this base being generated.
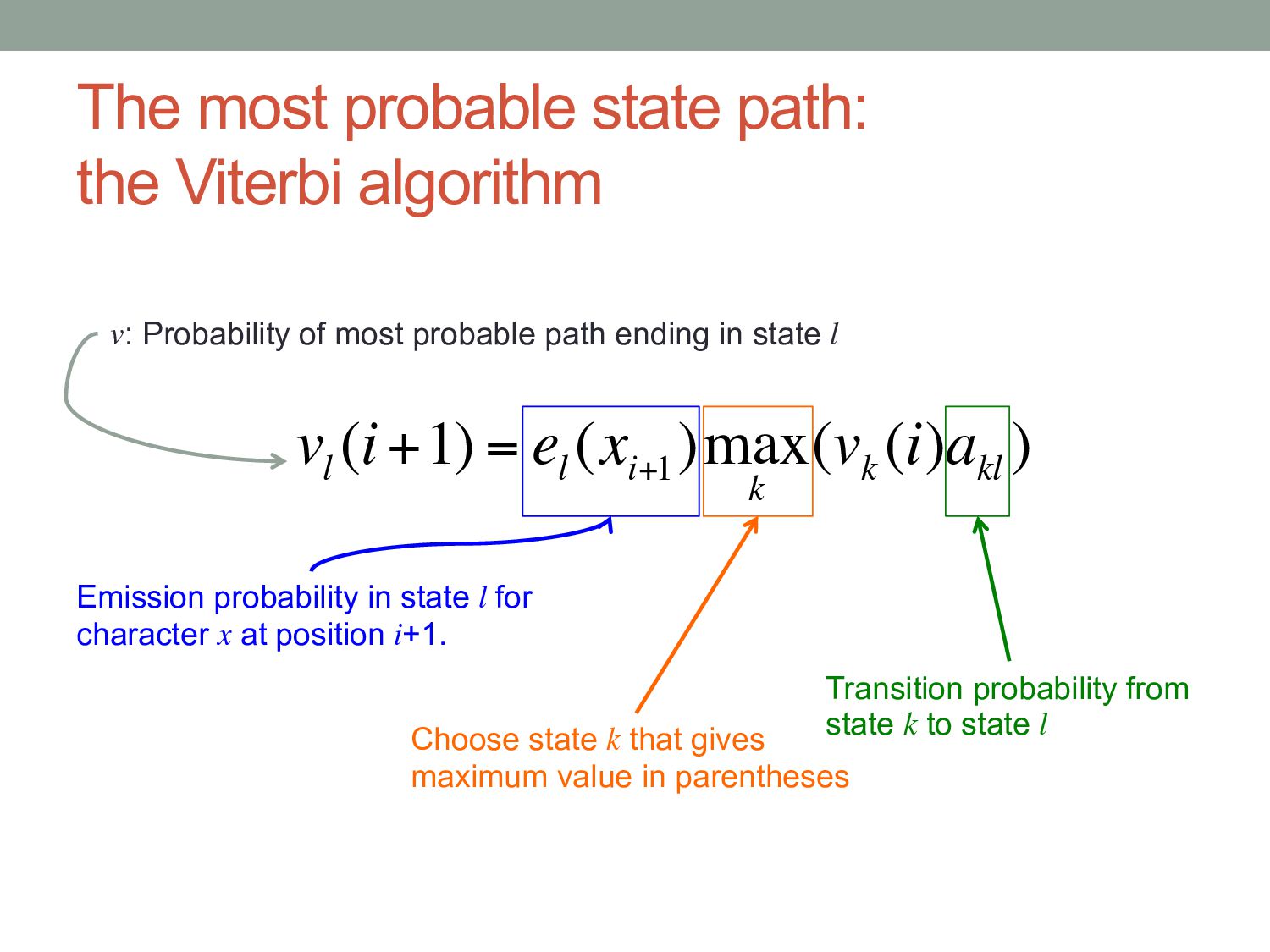
being in a state (l) in the next position (i + 1) on the sequence = Probability that state l produces the character at (i + 1) multiplied by the probability of being in state k at the current position multiplied by the transition probability of moving from state k to state l (where you choose k to give the highest overall probability, and k can be the same as l)
(i+1) = e l (x i+1 )max k (v k (i)a kl ) v: Probability of most probable path ending in state l Emission probability in state l for character x at position i+1. Transition probability from state k to state l Choose state k that gives maximum value in parentheses
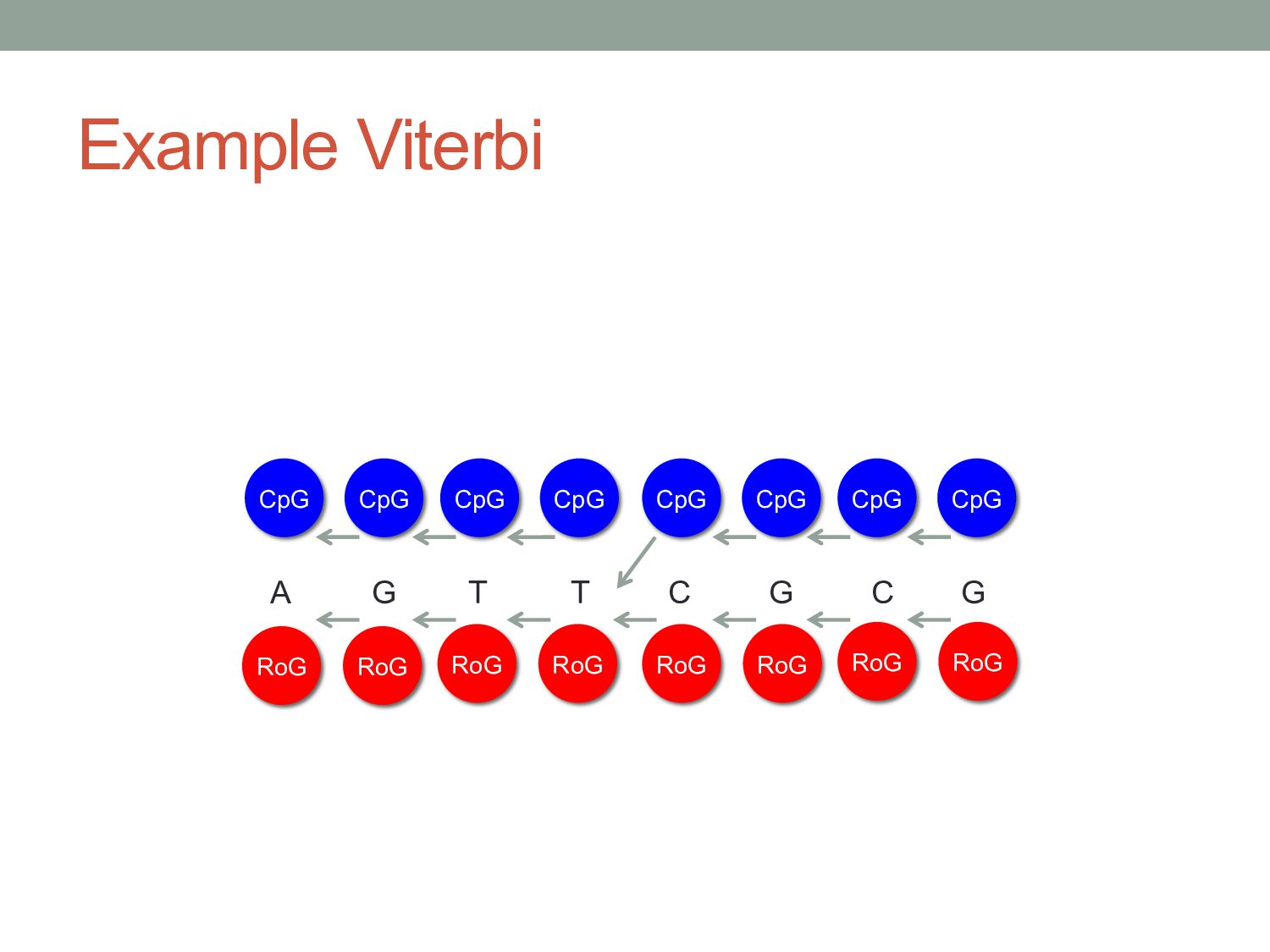
x = x1 ……xN Initialization: v0 (0) = 1 vk (0) = 0, for all k > 0 Iteration: for i = 1…L Traceback: for i = L…1 Statei-1 * = ptri (Statei *) v l (i+1) = e l (x i+1 )max k (v k (i)a kl ) ptr i (l) = argmax k (v k (i)a kl ) ß Pointer to state used in most probable path
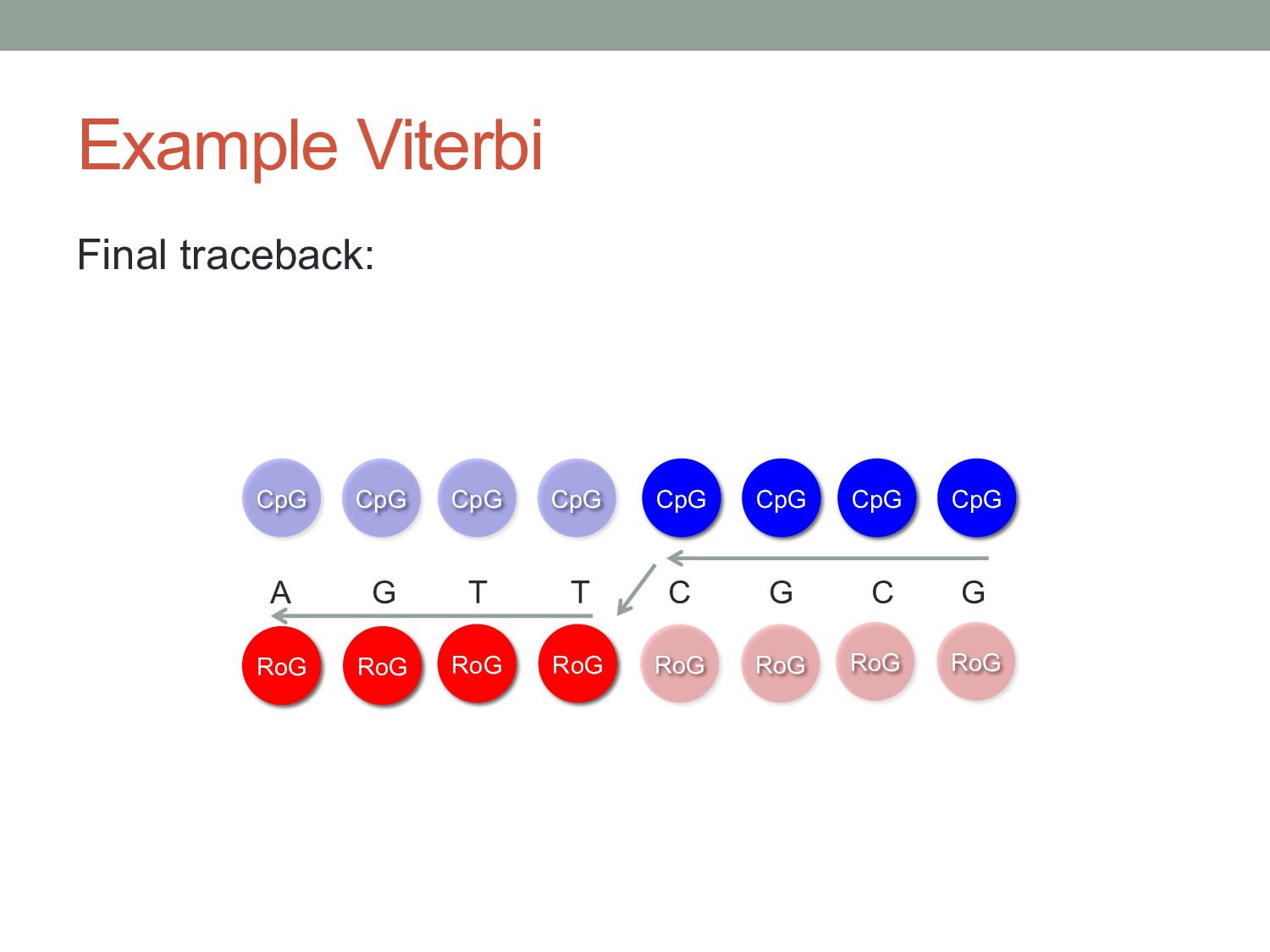
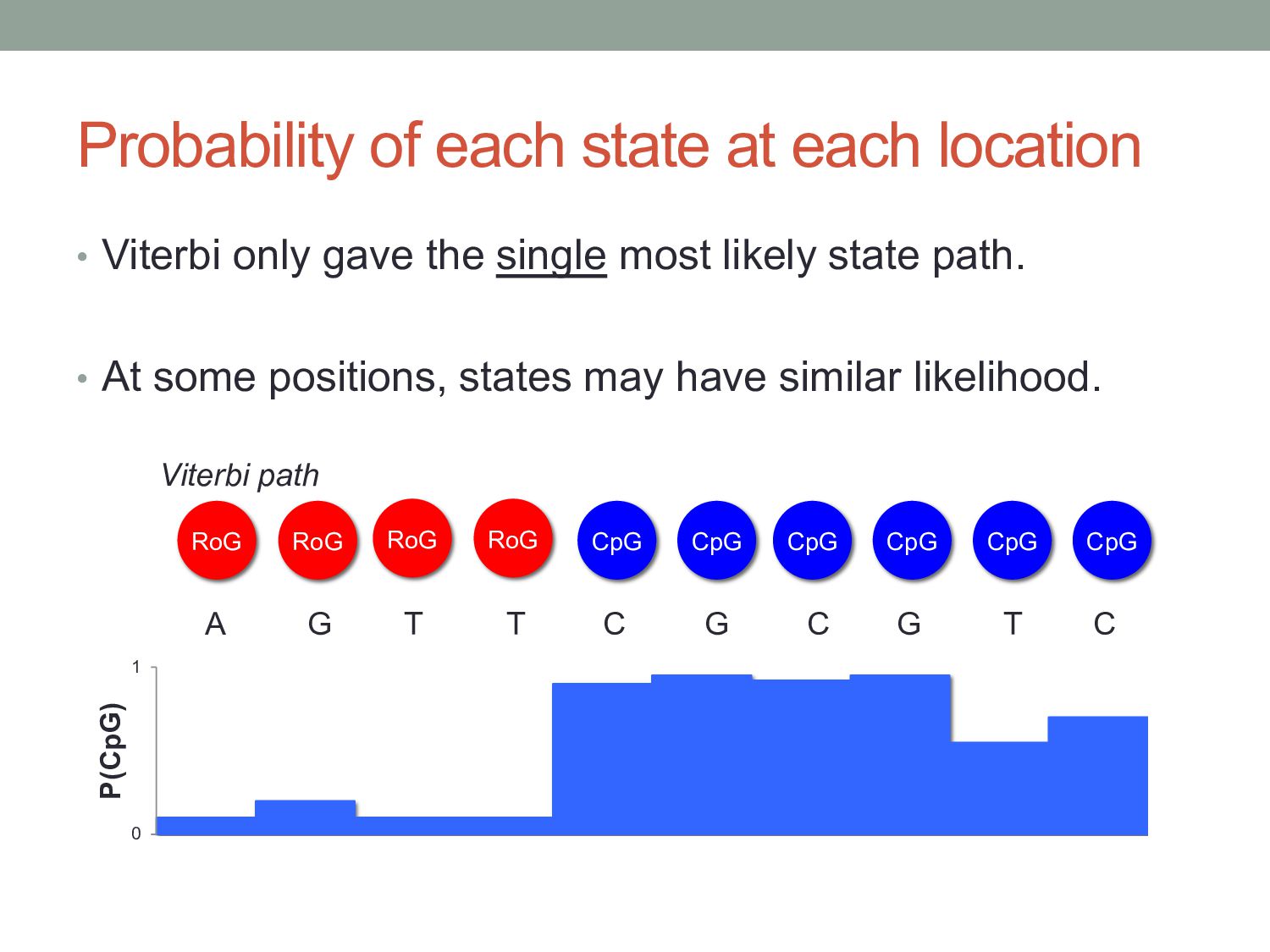
gave the single most likely state path. • At some positions, states may have similar likelihood. RoG A G T T C G C G RoG RoG RoG CpG CpG CpG CpG CpG CpG T C Viterbi path 0 1 P(CpG)
emission sequence, given the full HMM? • An observed sequence could be generated by many different state sequences. • Viterbi only gave the single most likely state sequence. • The Forward algorithm accounts for all possible ways in which an observed emission sequence could be generated.
= x1 ……xN Initialization: f0 (0) = 1 fk (0) = 0, for all k > 0 Iteration: for i = 1…L Termination: P(x) = Σk fk (N) ak0 f l (i+1) = e l (x i+1 ) ( f k (i)a kl ) k ∑ Sum instead of choosing max
care about the probabilities of the sequences? • Forward algorithm gives the probability of state k given everything that has come before. • Backward algorithm gives the probability of state k given everything that comes after. P(State i = k | x) = f k (i)b k (i) P(x) “Posterior” probability: probability of seeing state k at position i Think of this as a weighted labeling
What do we need? • Emission probabilities: frequencies of each k-mer in locations with each state label. • Transition probabilities: how often do we see one state flip into another? • Easy if we have a labeled training set. • If we don’t … can we find parameters from the data? • Machine-learning! • Baum-Welch algorithm – instance of Expectation Maximization
• Initialization: • Pick arbitrary HMM parameters. • Iteration: Calculate all fk (i) using forward algorithm Calculate all bk (i) using backward algorithm Calculate new model parameters given the weighted state labels • Termination: Stop if the HMM parameters stop changing between iterations
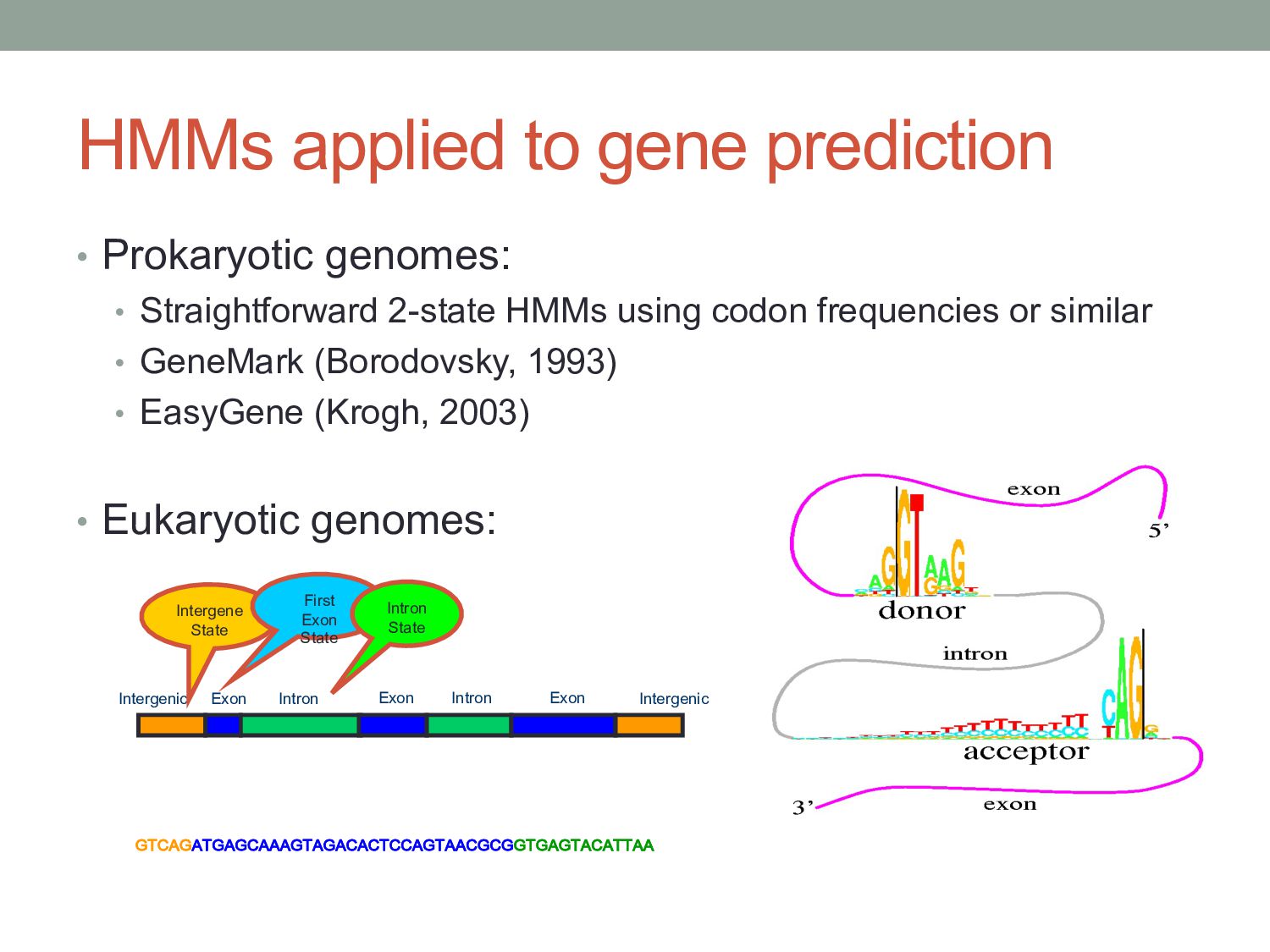
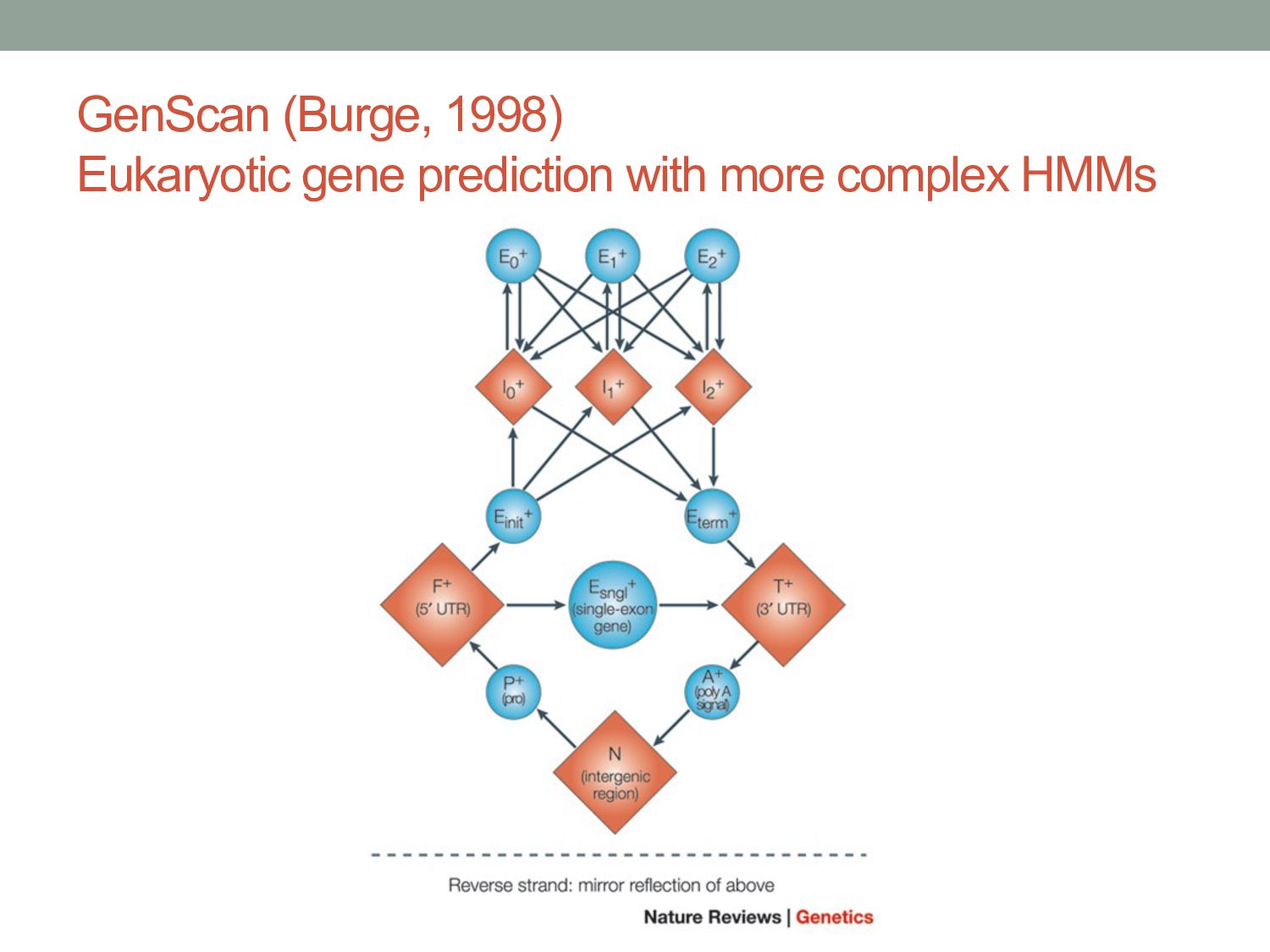
2-state HMMs using codon frequencies or similar • GeneMark (Borodovsky, 1993) • EasyGene (Krogh, 2003) • Eukaryotic genomes: !"#$!$"!$!#$$$!"$!$#$#"##$!"$$#!#!!"!$!"$#$""$$ %&'( %&'( %&'( )(*+'( )(*+'( )(*,+-,(./ )(*,+-,(./ Intergene State First Exon State Intron State
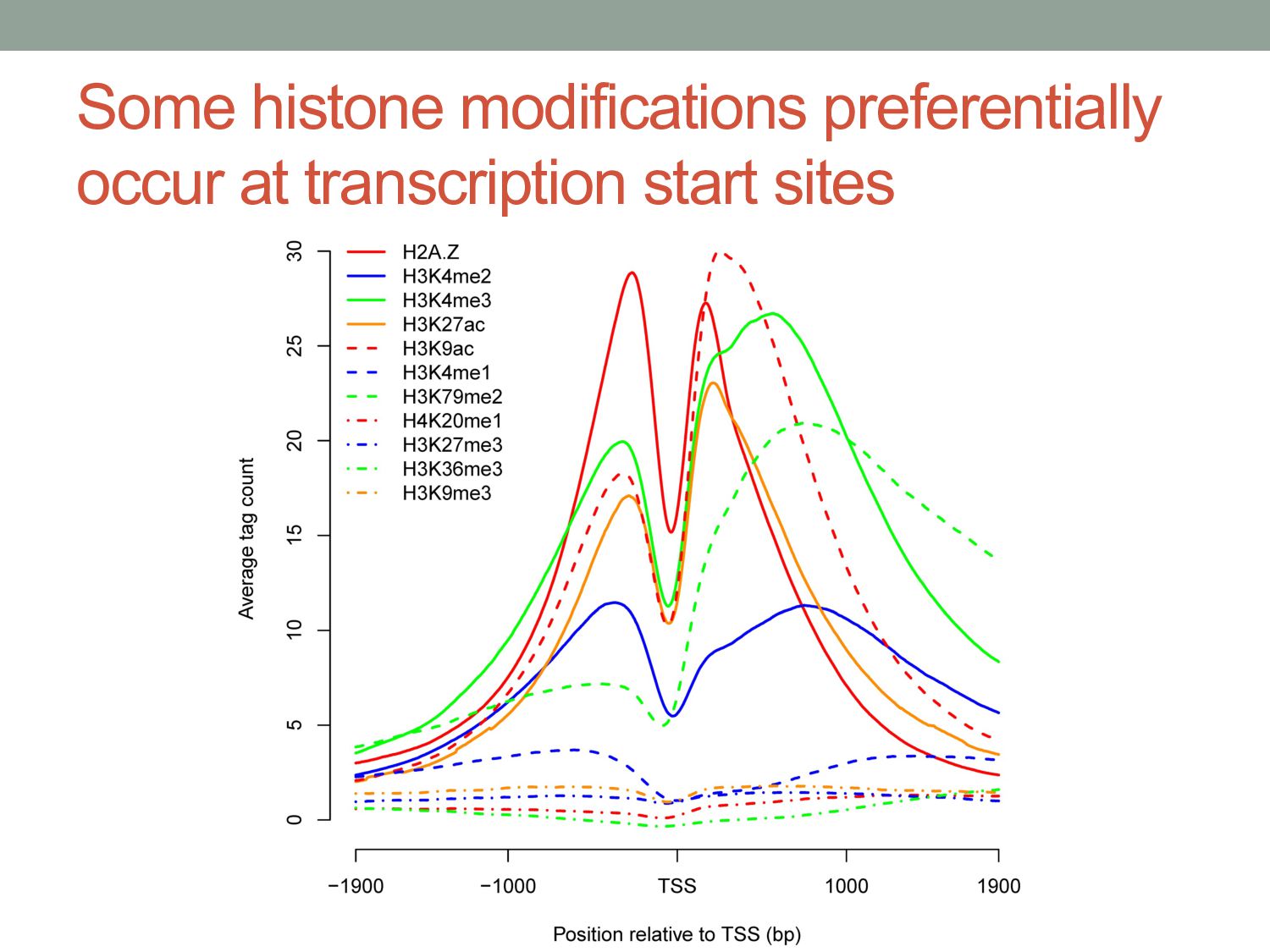
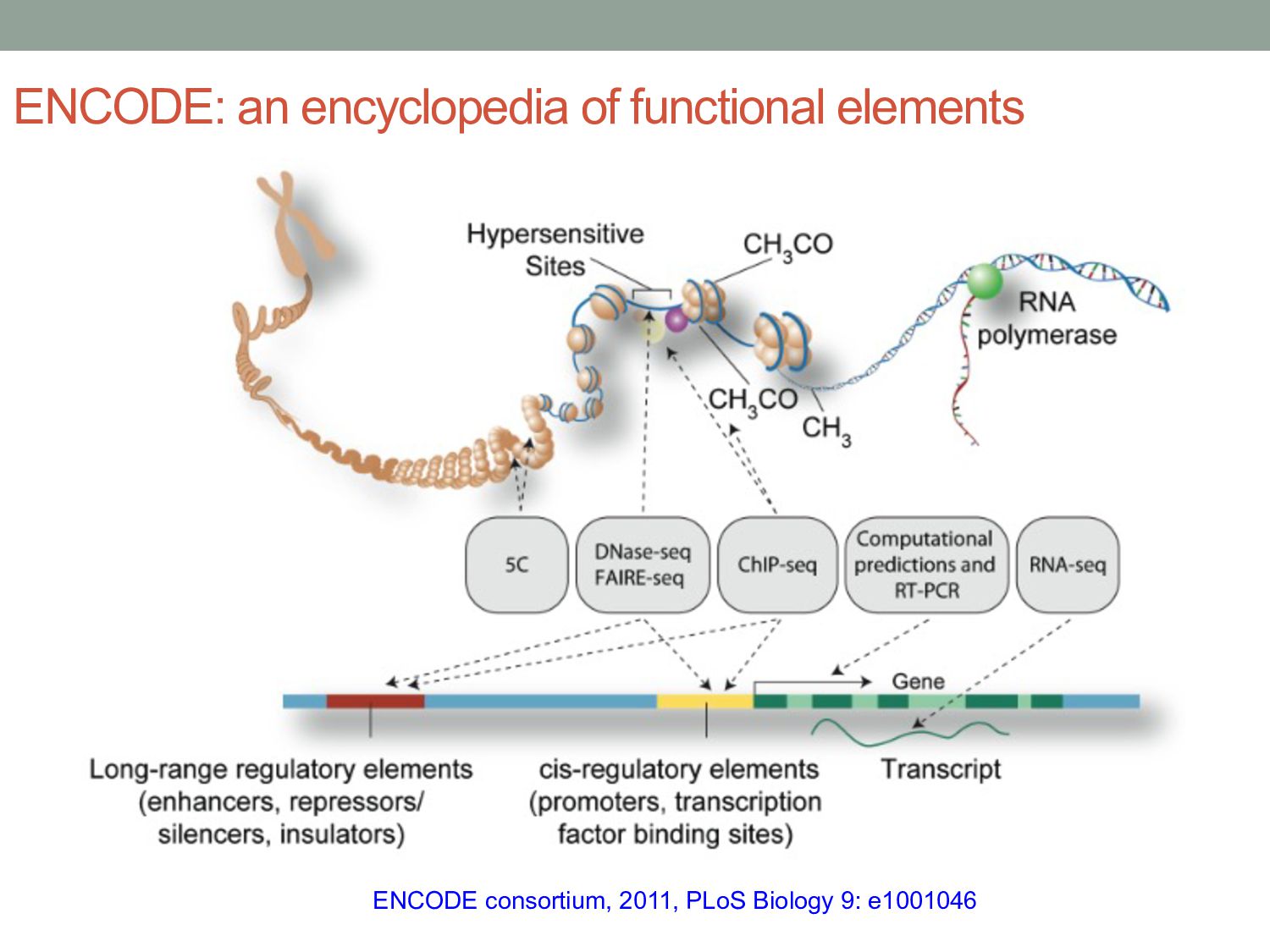
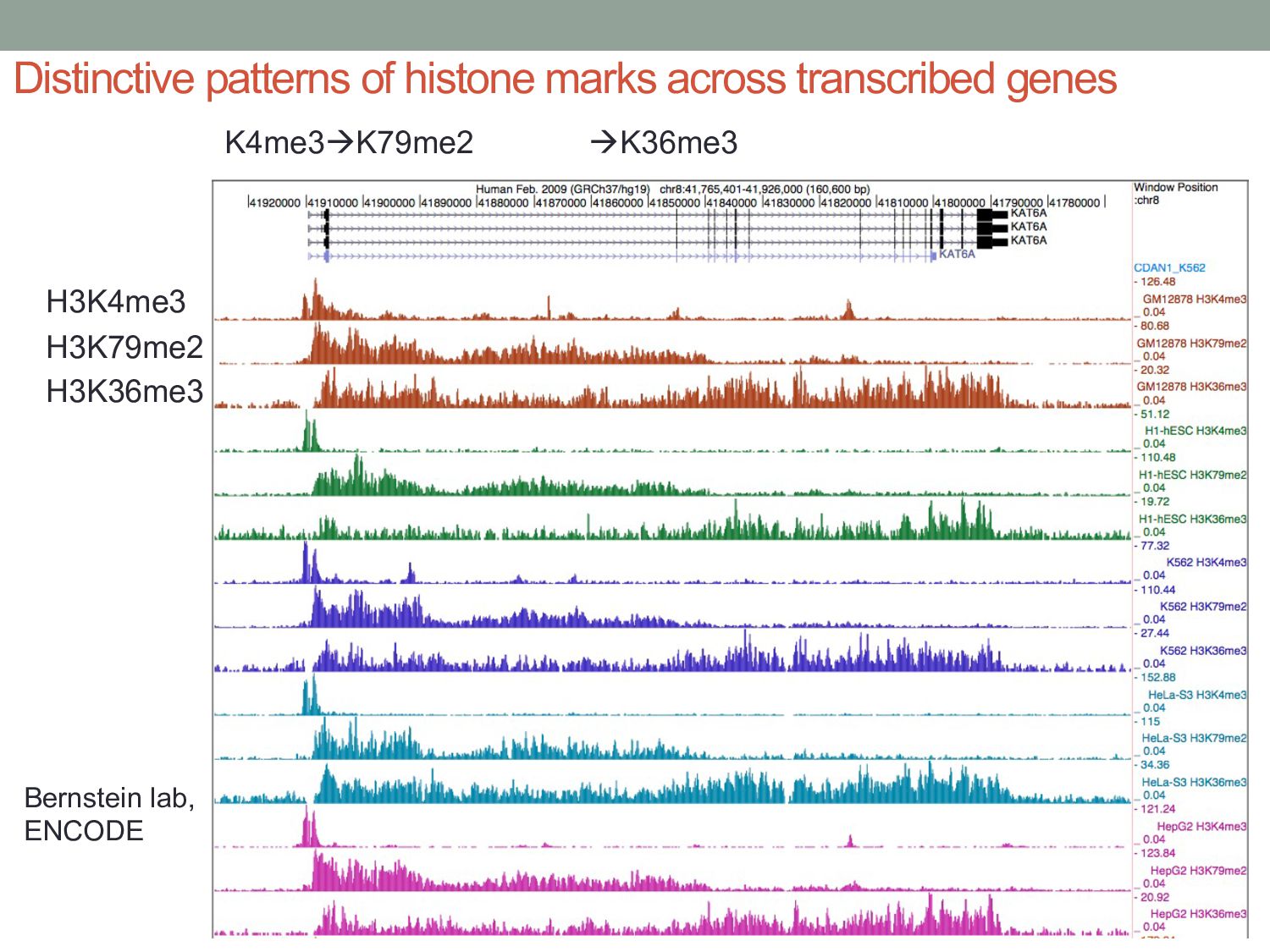
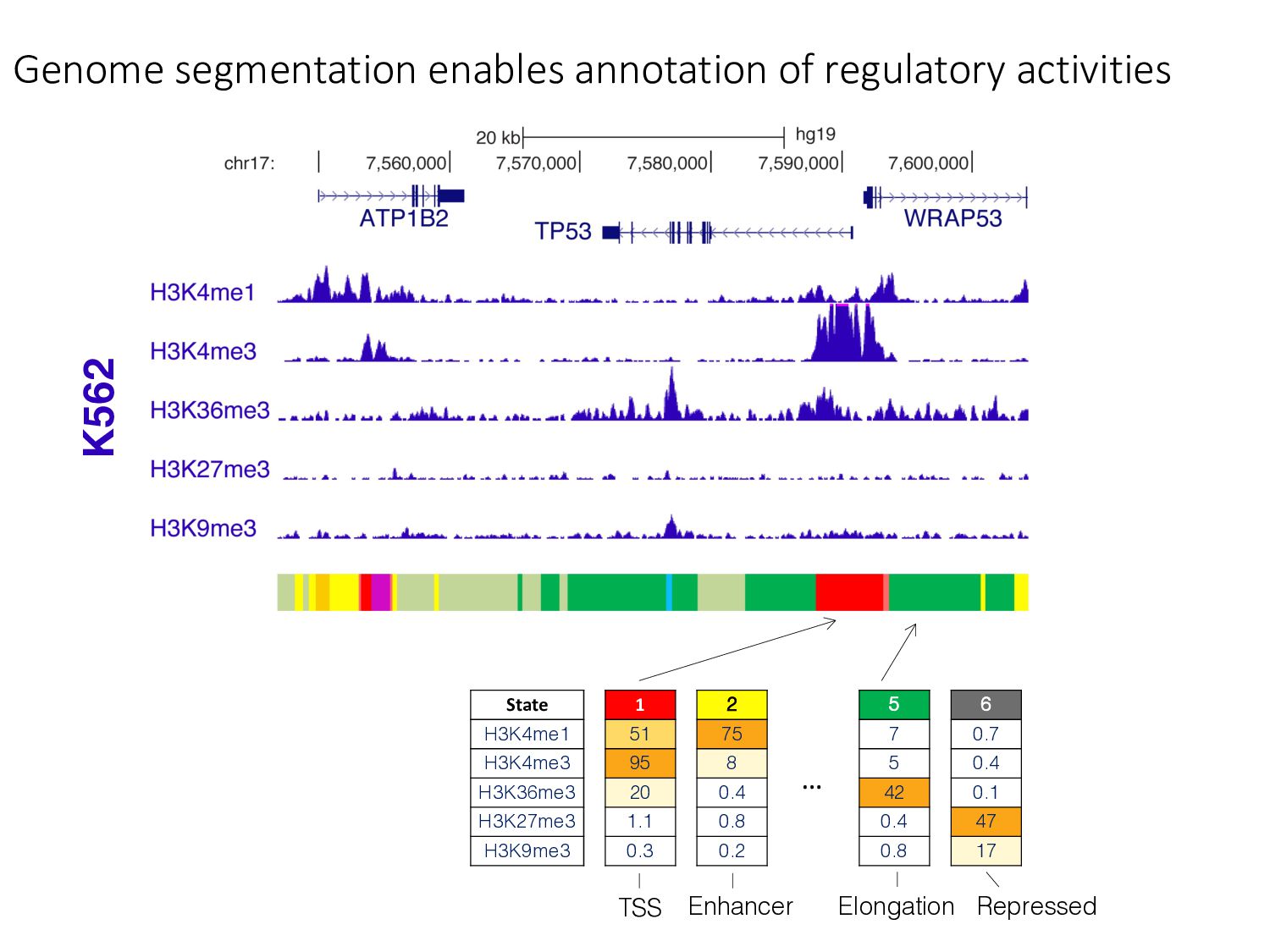
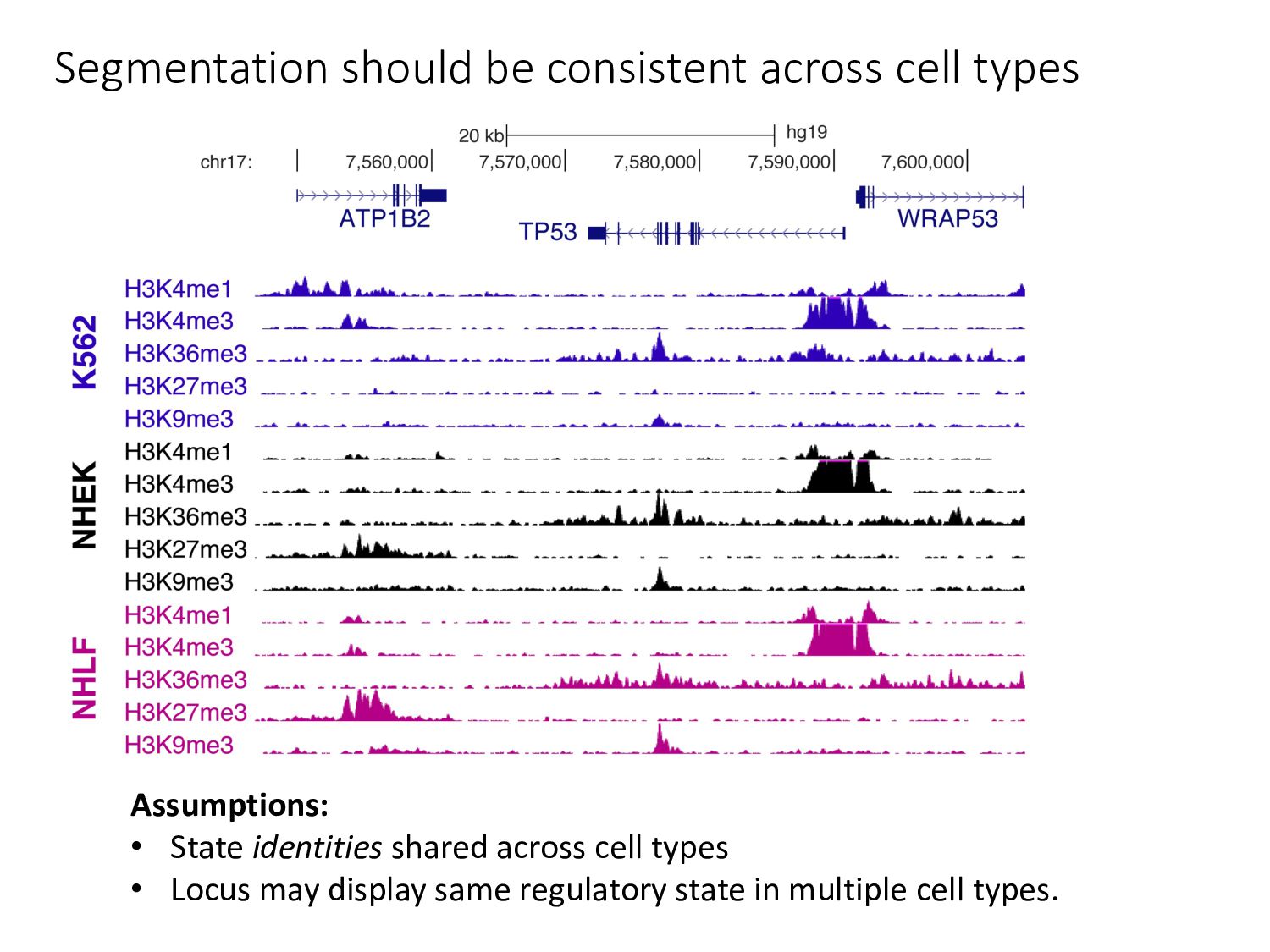
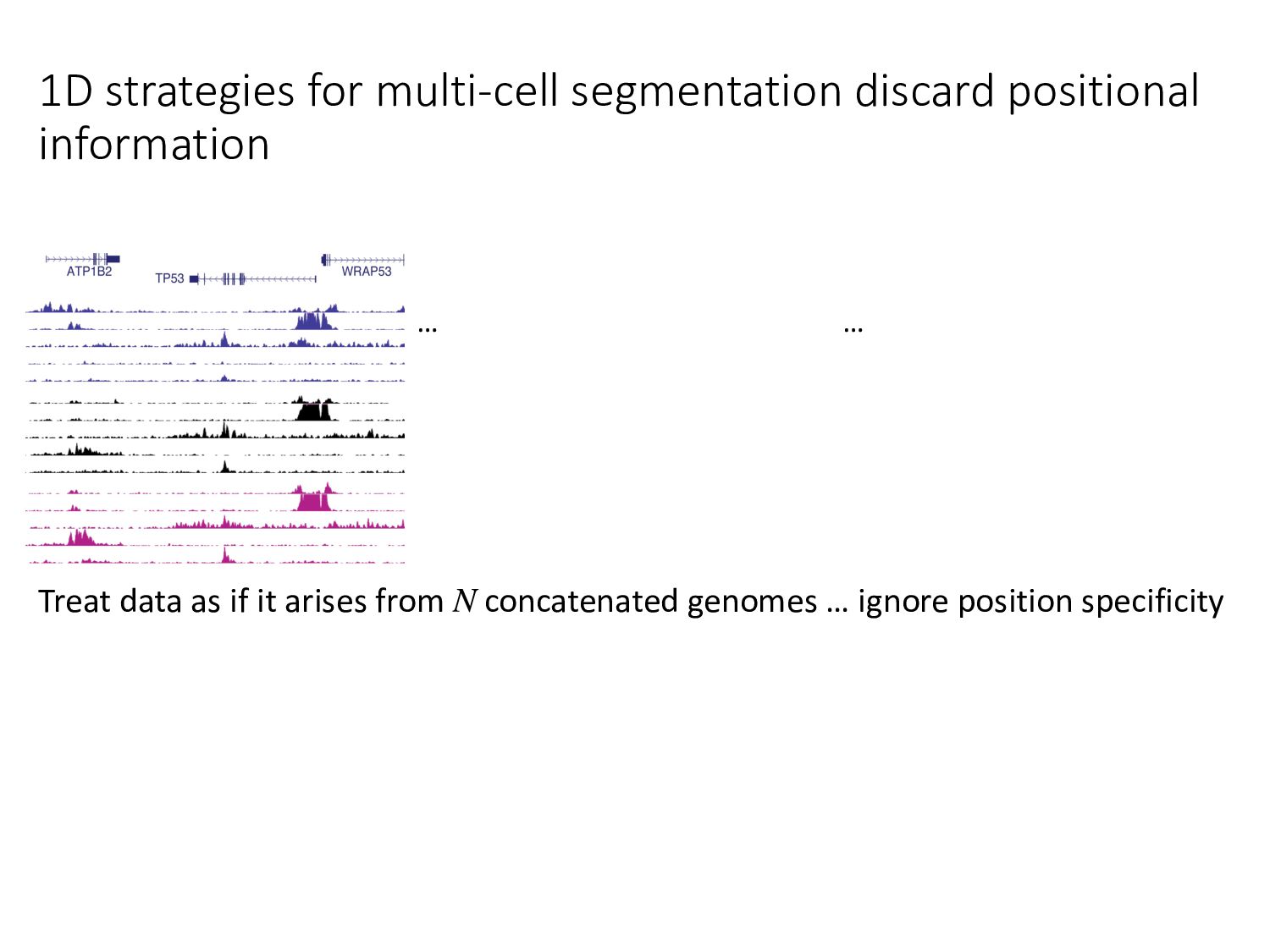
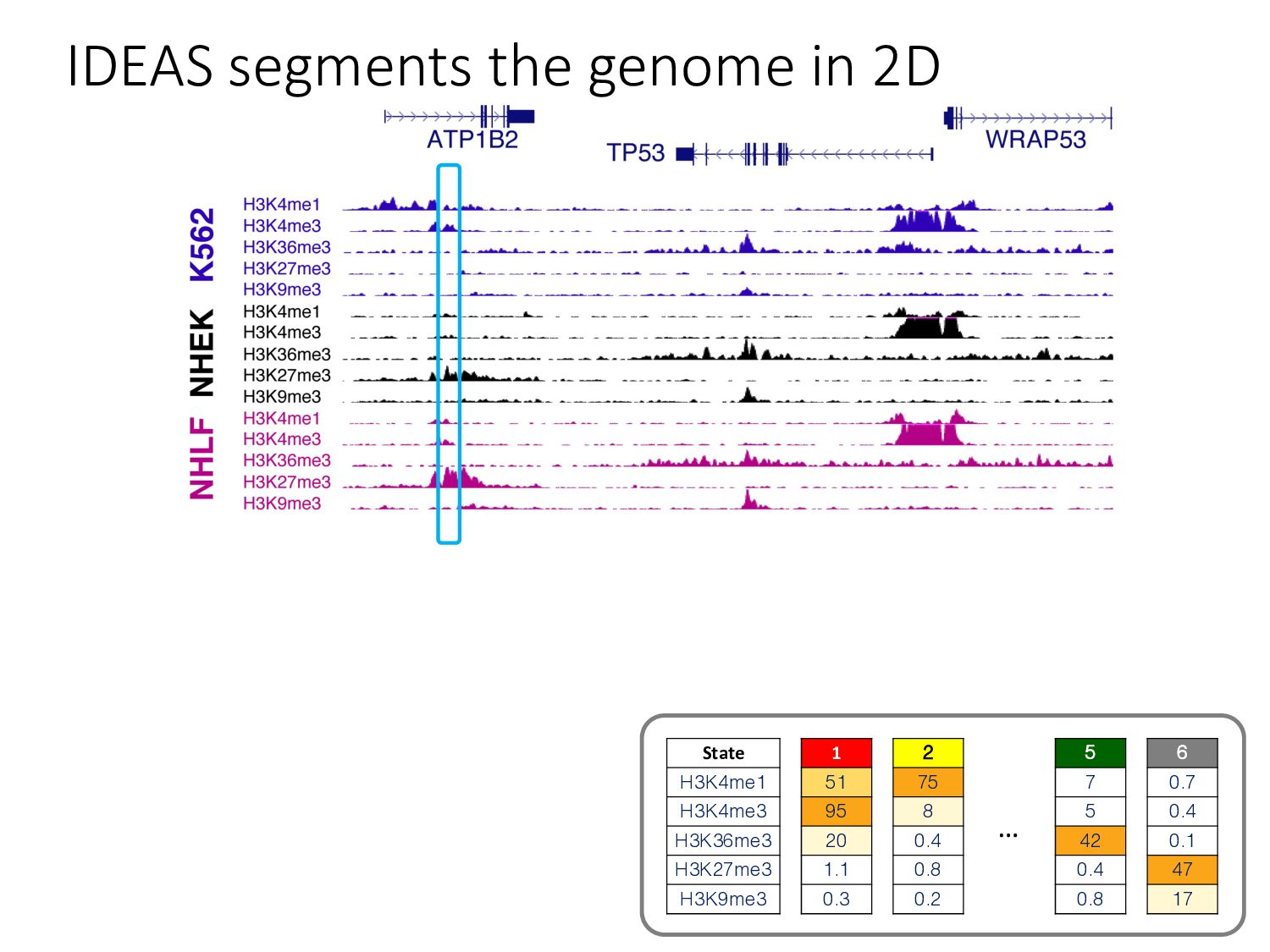
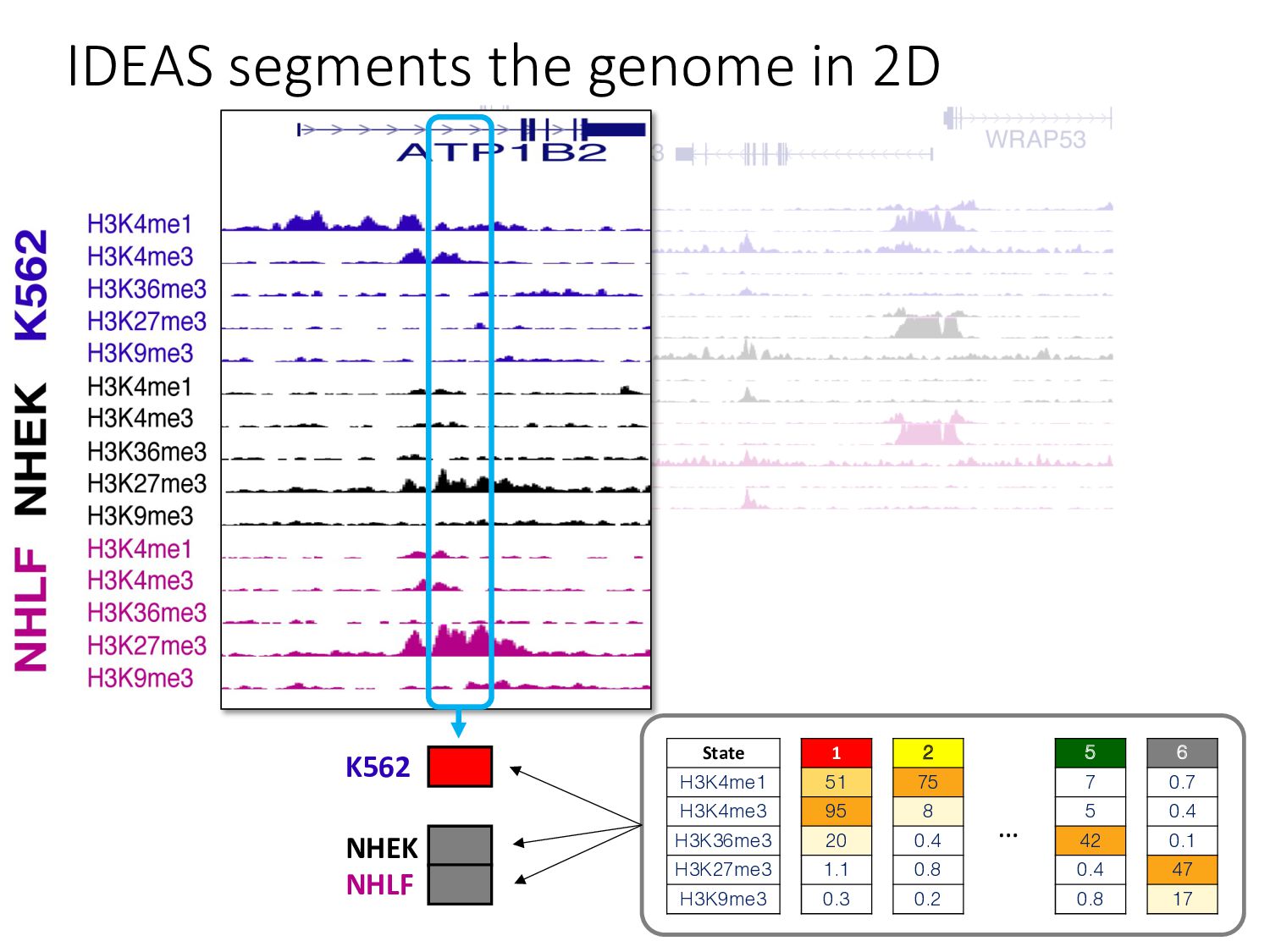
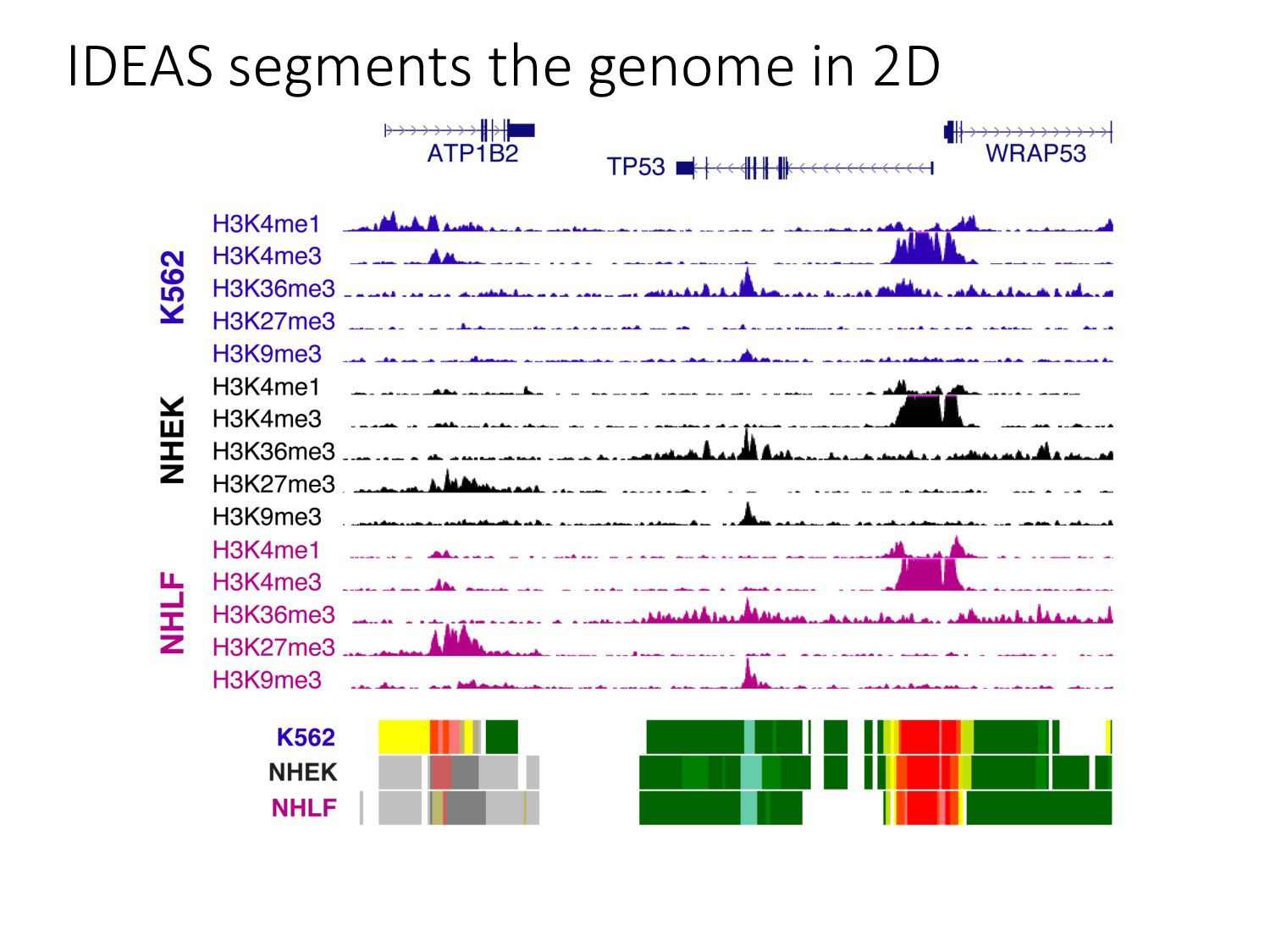
particular combination of epigenomic activities appearing at the same genomic loci. • Patterns over combinations of: • Histone modifications • Protein-DNA binding • RNA polymerase • DNaseI accessibility • DNA methylation • etc... • Chromatin states correspond to particular modes of functional activity at the underlying regions of the genome.
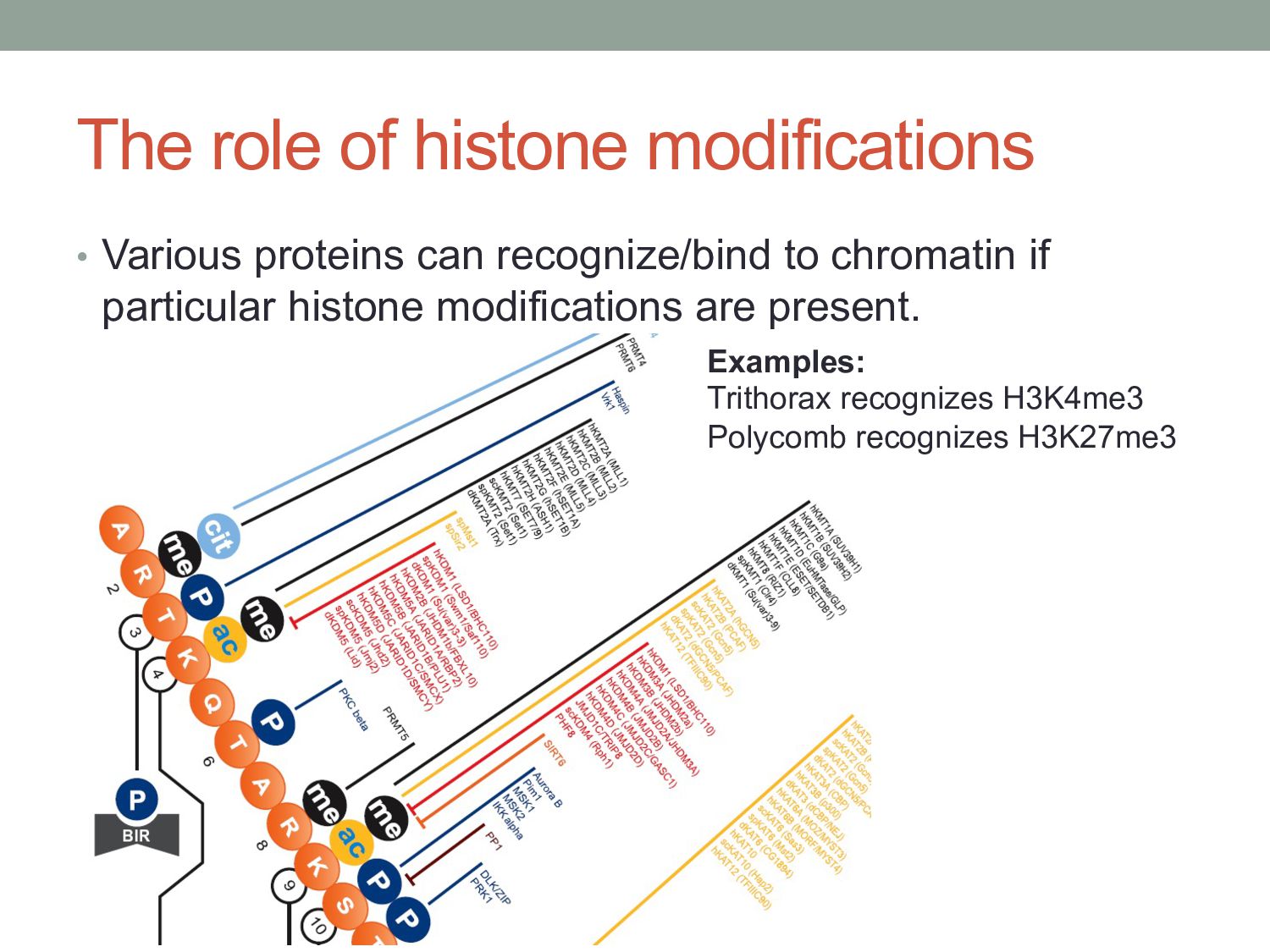
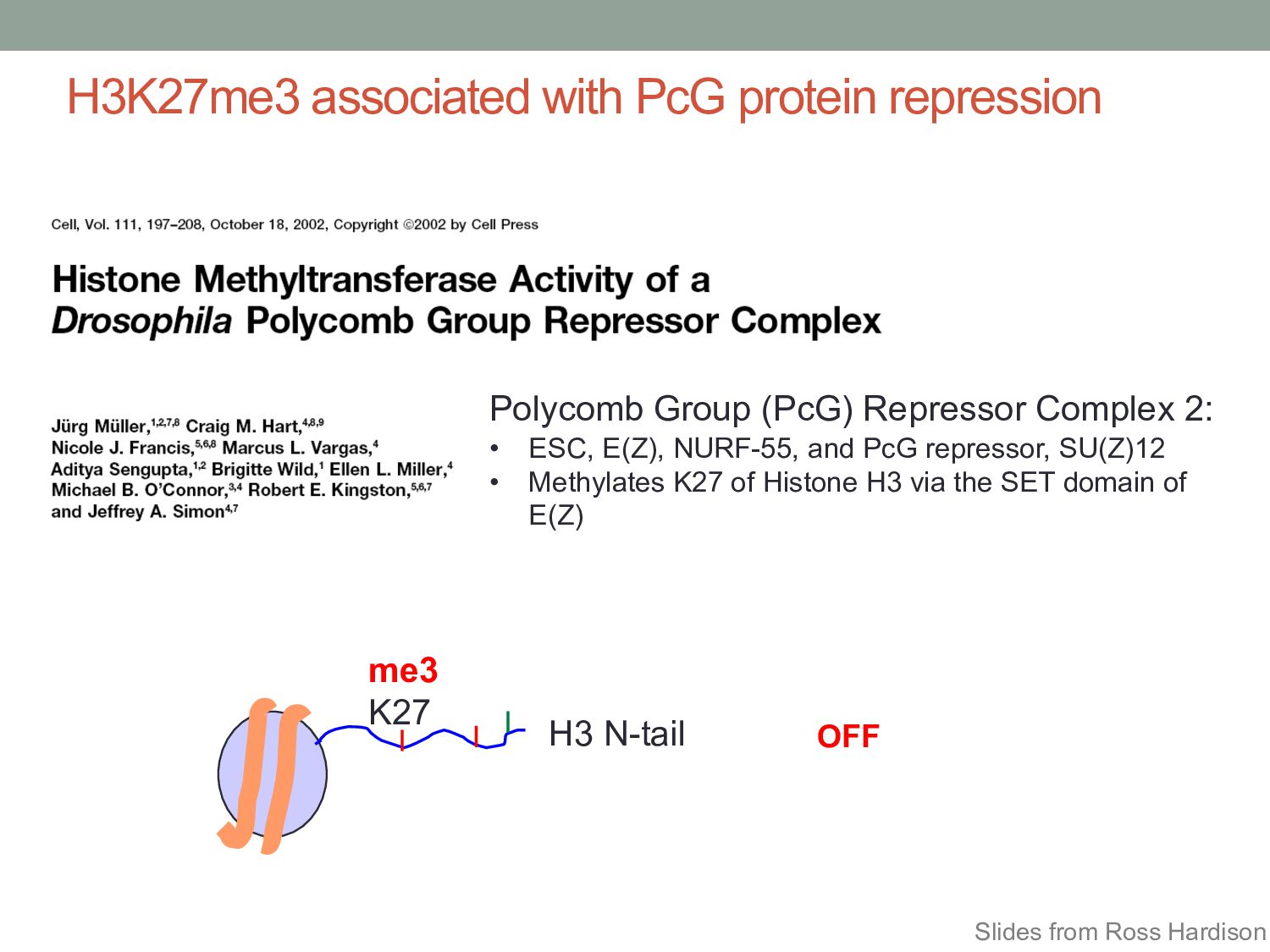
Complex 2: • ESC, E(Z), NURF-55, and PcG repressor, SU(Z)12 • Methylates K27 of Histone H3 via the SET domain of E(Z) me3 H3 N-tail K27 OFF Slides from Ross Hardison
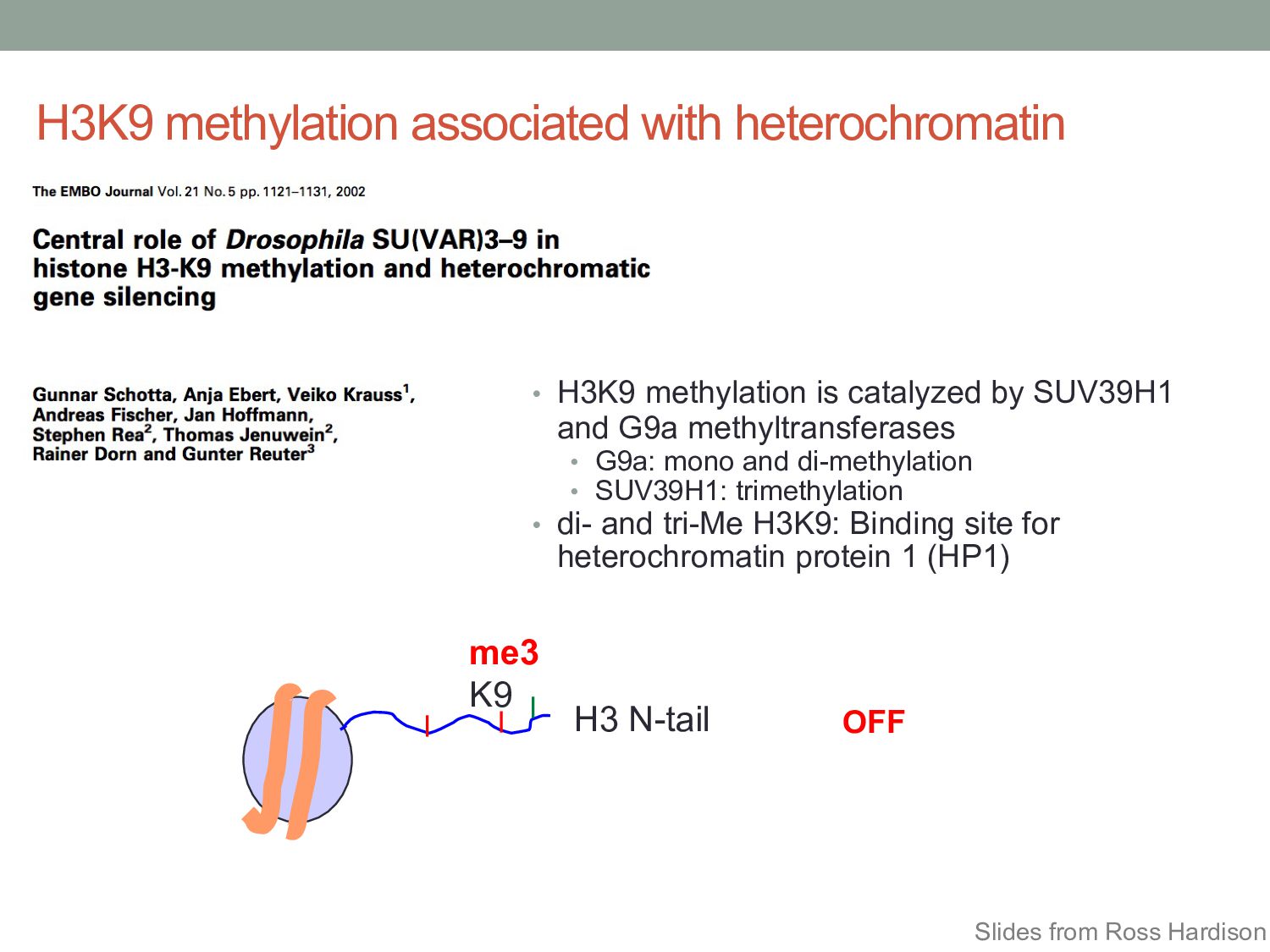
• H3K9 methylation is catalyzed by SUV39H1 and G9a methyltransferases • G9a: mono and di-methylation • SUV39H1: trimethylation • di- and tri-Me H3K9: Binding site for heterochromatin protein 1 (HP1) Slides from Ross Hardison
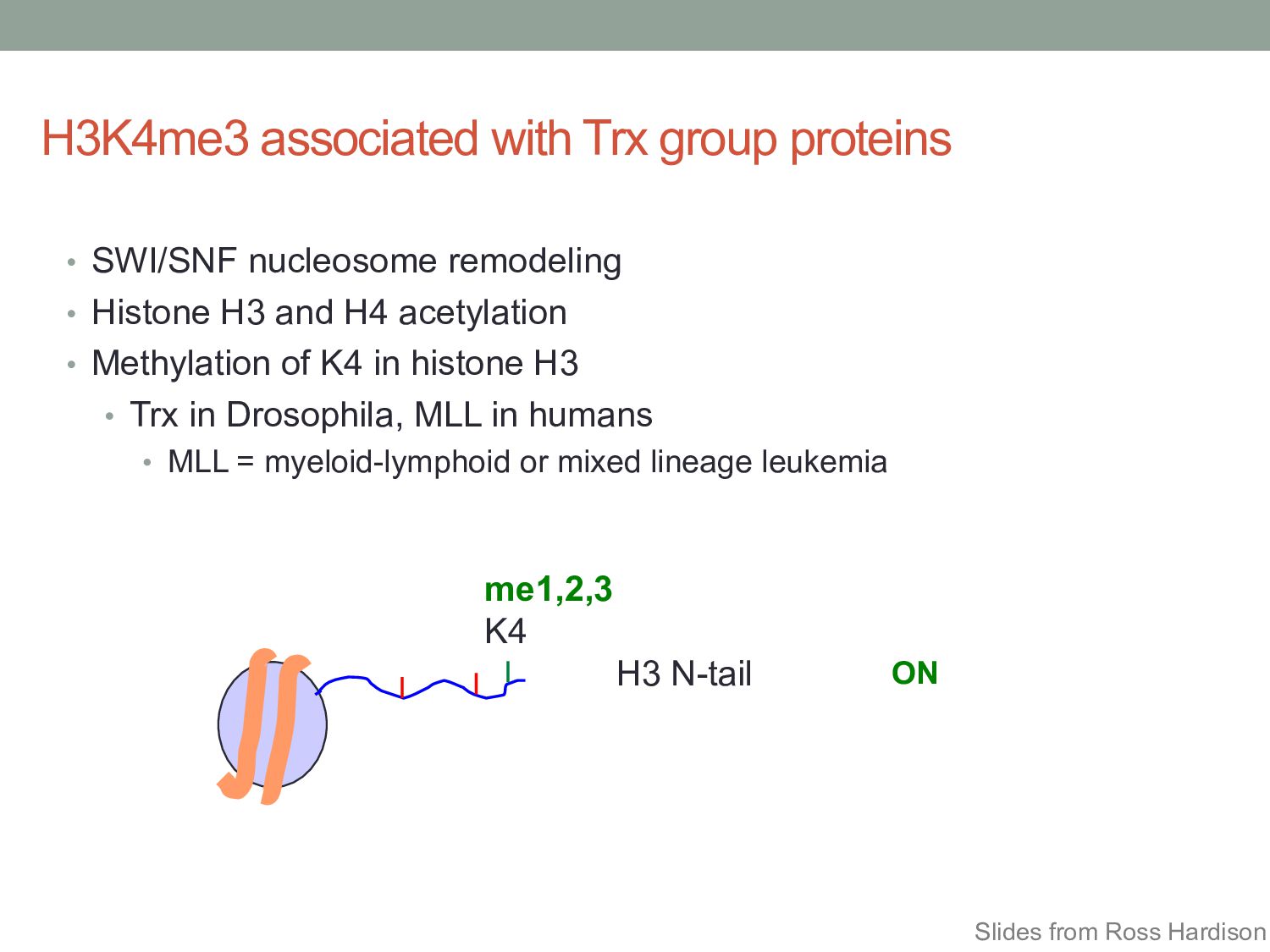
• Histone H3 and H4 acetylation • Methylation of K4 in histone H3 • Trx in Drosophila, MLL in humans • MLL = myeloid-lymphoid or mixed lineage leukemia H3 N-tail me1,2,3 K4 ON Slides from Ross Hardison
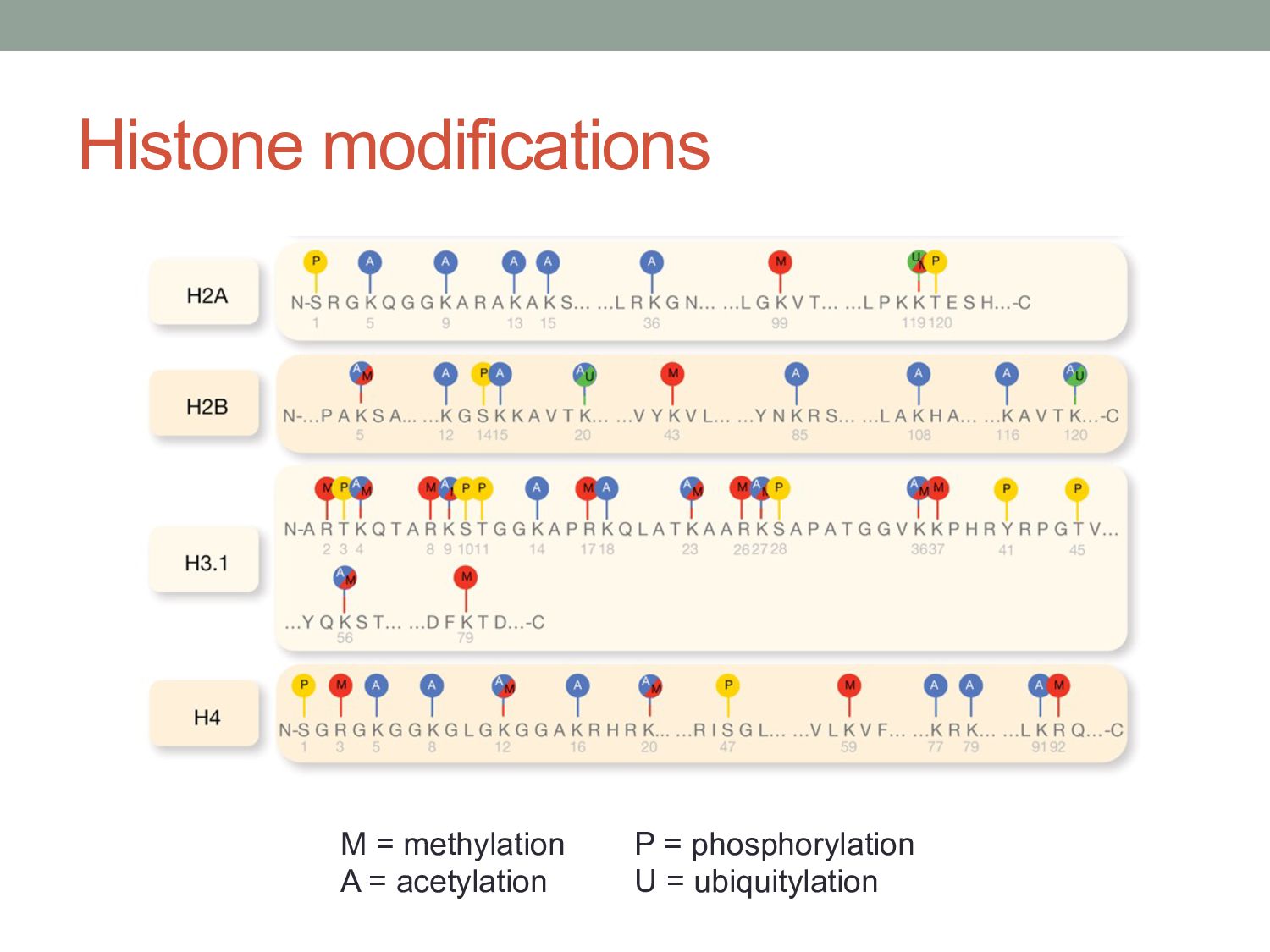
exist that extends the information potential of DNA. • Histone modifications and chromatin-associated proteins might serve to store information about the regulatory ‘state’ of underlying genes, and might allow this information to be stably inherited by offspring cells. • Potential complexity of the code is enormous: • e.g. 19 lysine residues on H3 which can be un-, mono-, di-, or tri- methylated… so 419 possible combinations?
there? • How do chromatin states change across cell types? • What do chromatin states tell us about human genetics & health? • Study variants in functional regions as annotated by HMMs using GWAS. • Examine how chromatin state changes between normal & disease tissues.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}