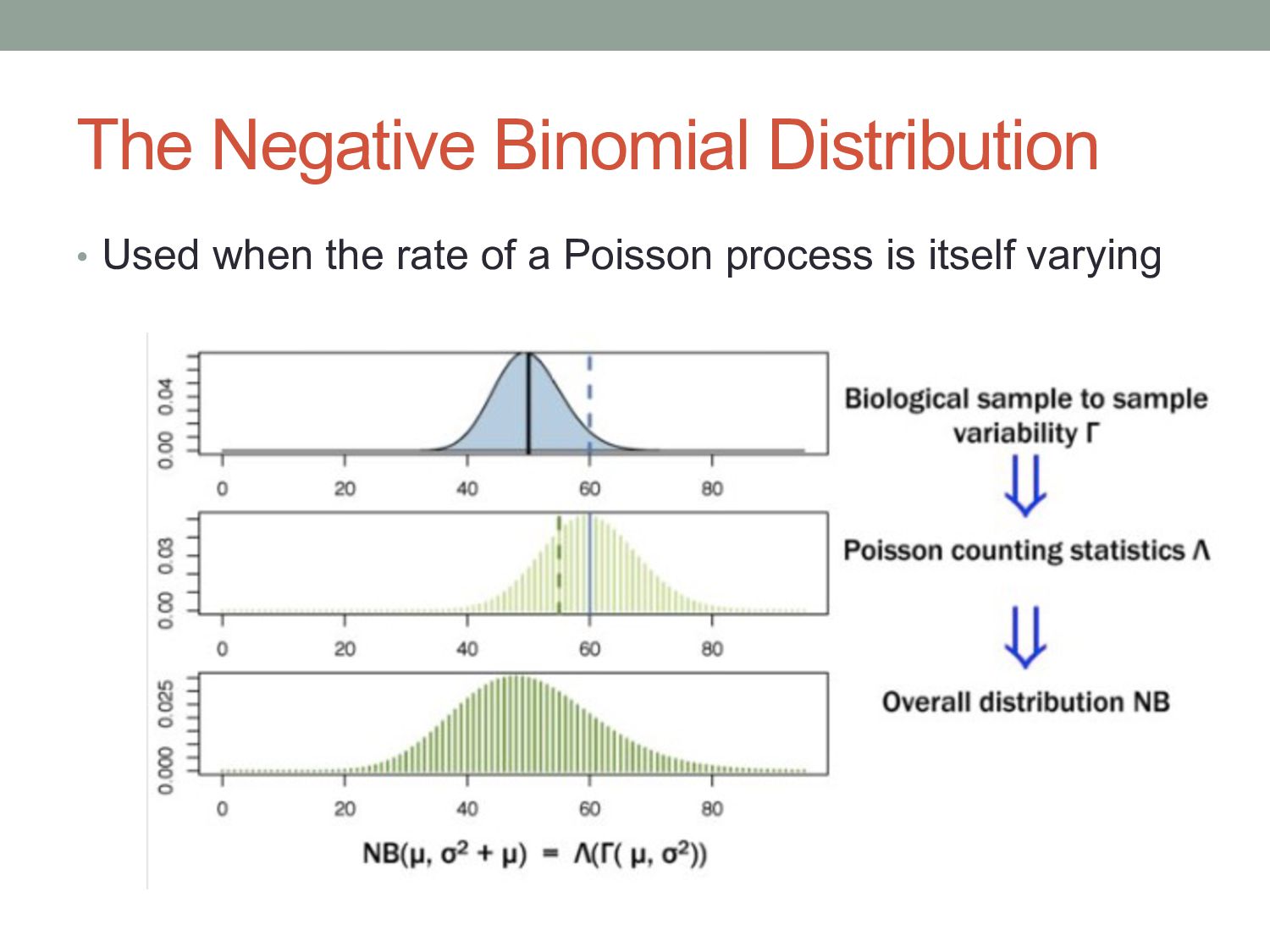
i in conditionx depends on a Negative Binomial (NB) function. • The mean of the NB depends on the concentration of mRNA fragments for gene i in condition x. • The variance of the NB is a combination of two effects: • “Shot noise”; equal to the mean of the NB – i.e., the Poisson effect. + • Biological variance between replicates. • We estimate the NB variance parameter by observing how variable the counts are across biological replicates.
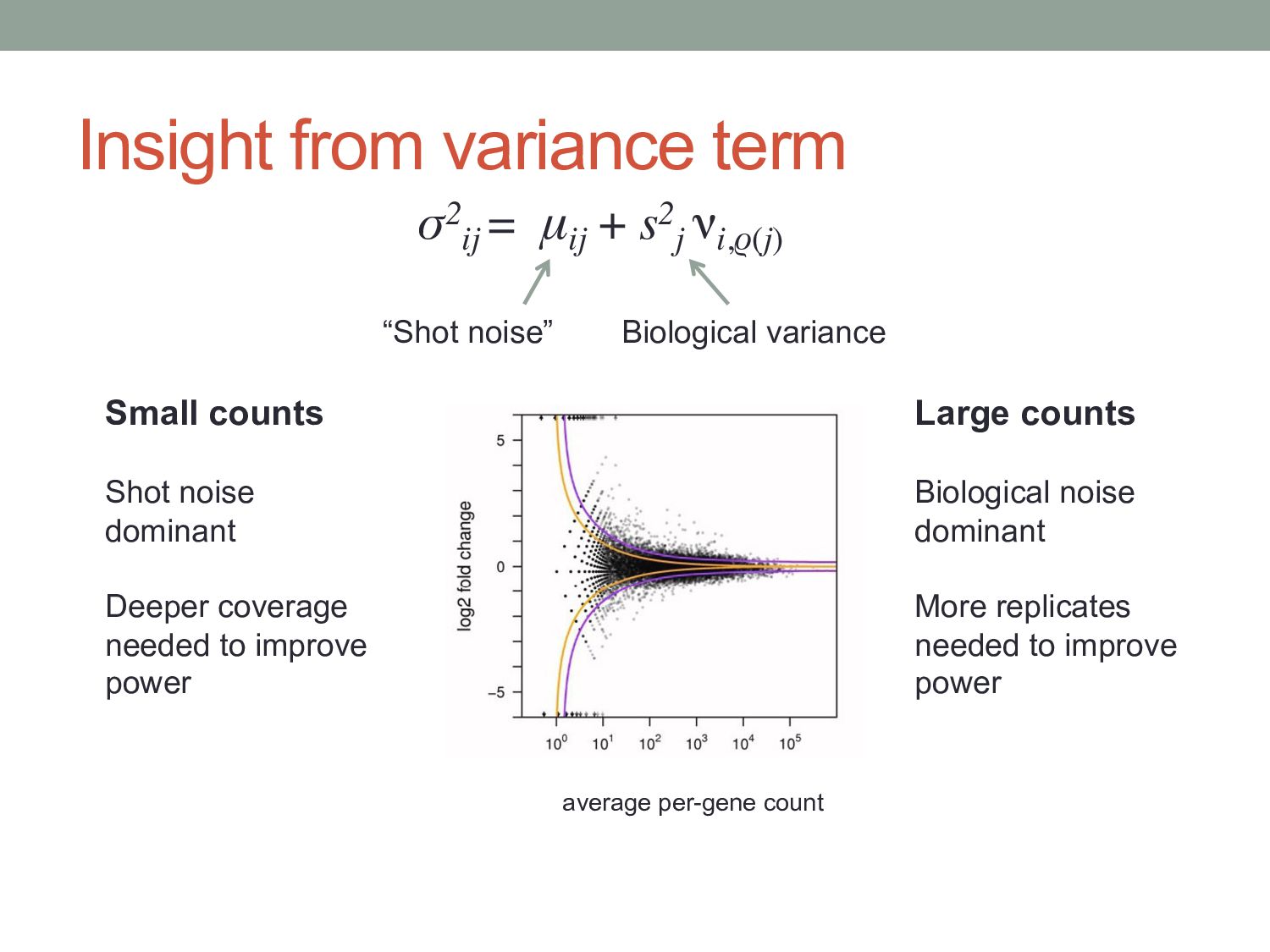
) i = gene j = sample kij = count for gene i in sample j qi,ρ(j) = expected concentration of fragments of gene i in condition ρ(j) sj = scaling factor νi,ρ(j) = per gene variance estimated from smooth function of q Where: μij =qi,ρ(j) sj σ2 ij = μij + s2 j νi,ρ(j) “Shot noise” Biological variance between replicates mean: variance:
μij + s2 j νi,ρ(j) “Shot noise” Biological variance Small counts Shot noise dominant Deeper coverage needed to improve power Large counts Biological noise dominant More replicates needed to improve power
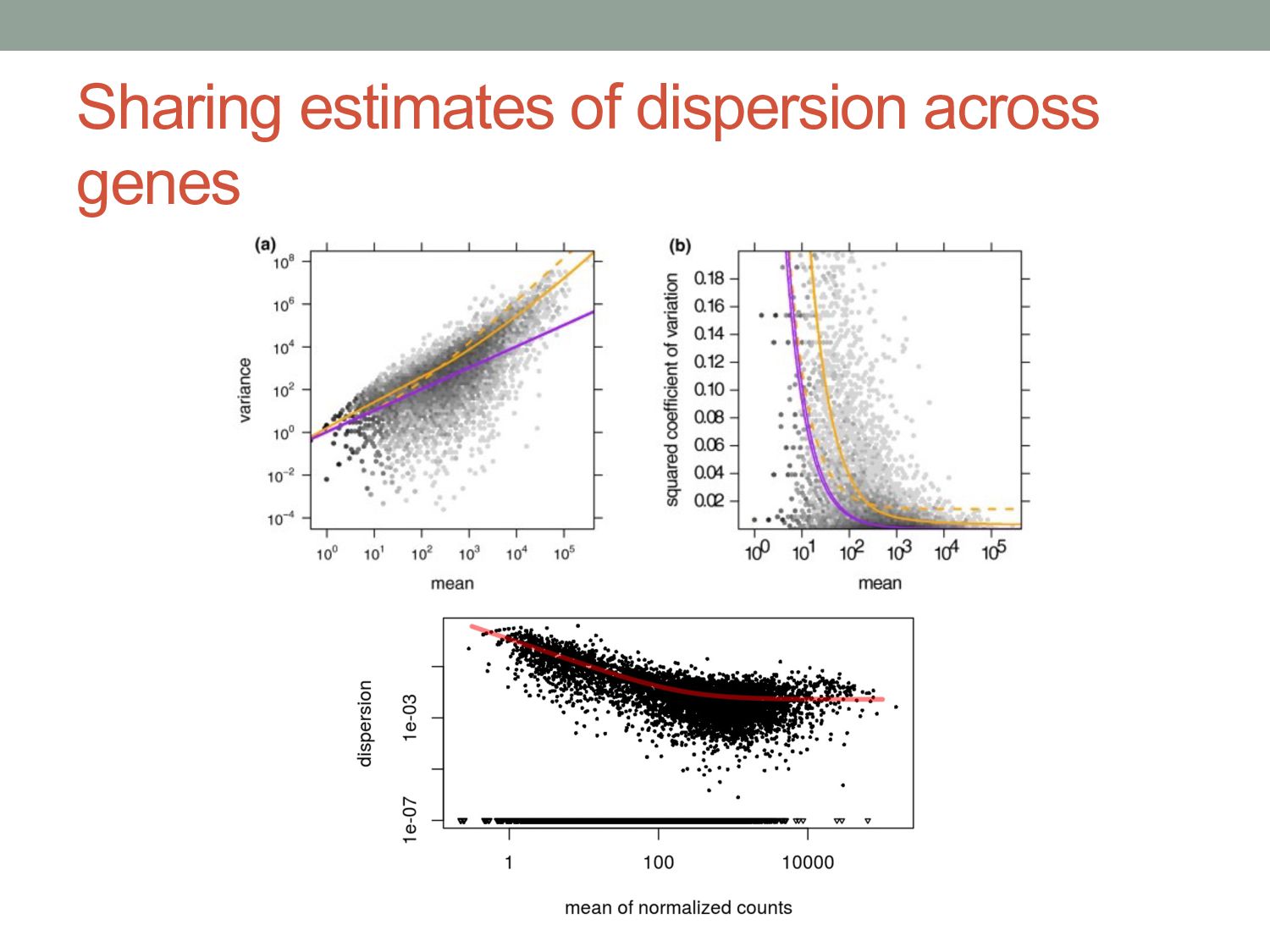
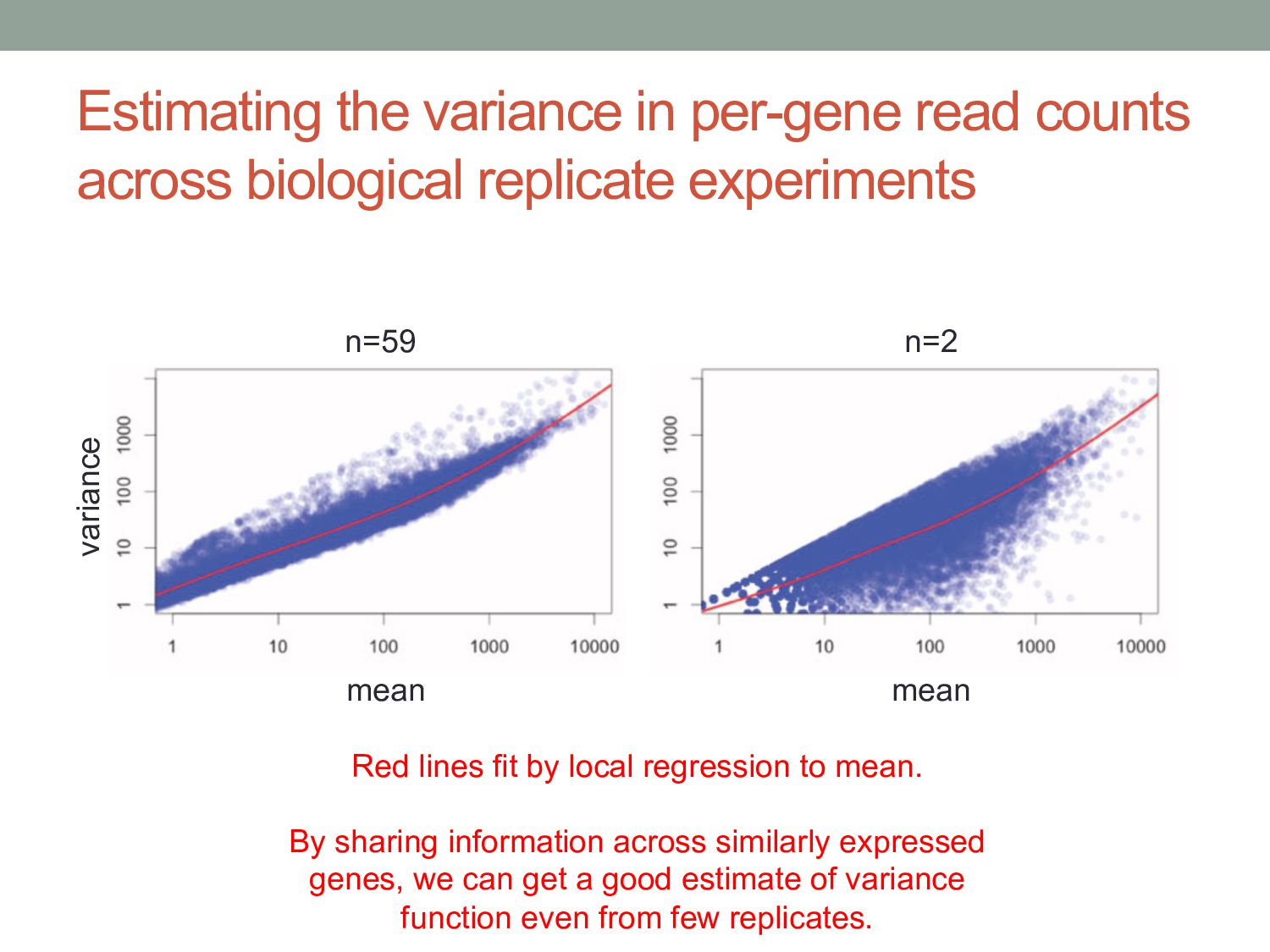
experiments n=59 n=2 mean mean variance Red lines fit by local regression to mean. By sharing information across similarly expressed genes, we can get a good estimate of variance function even from few replicates.
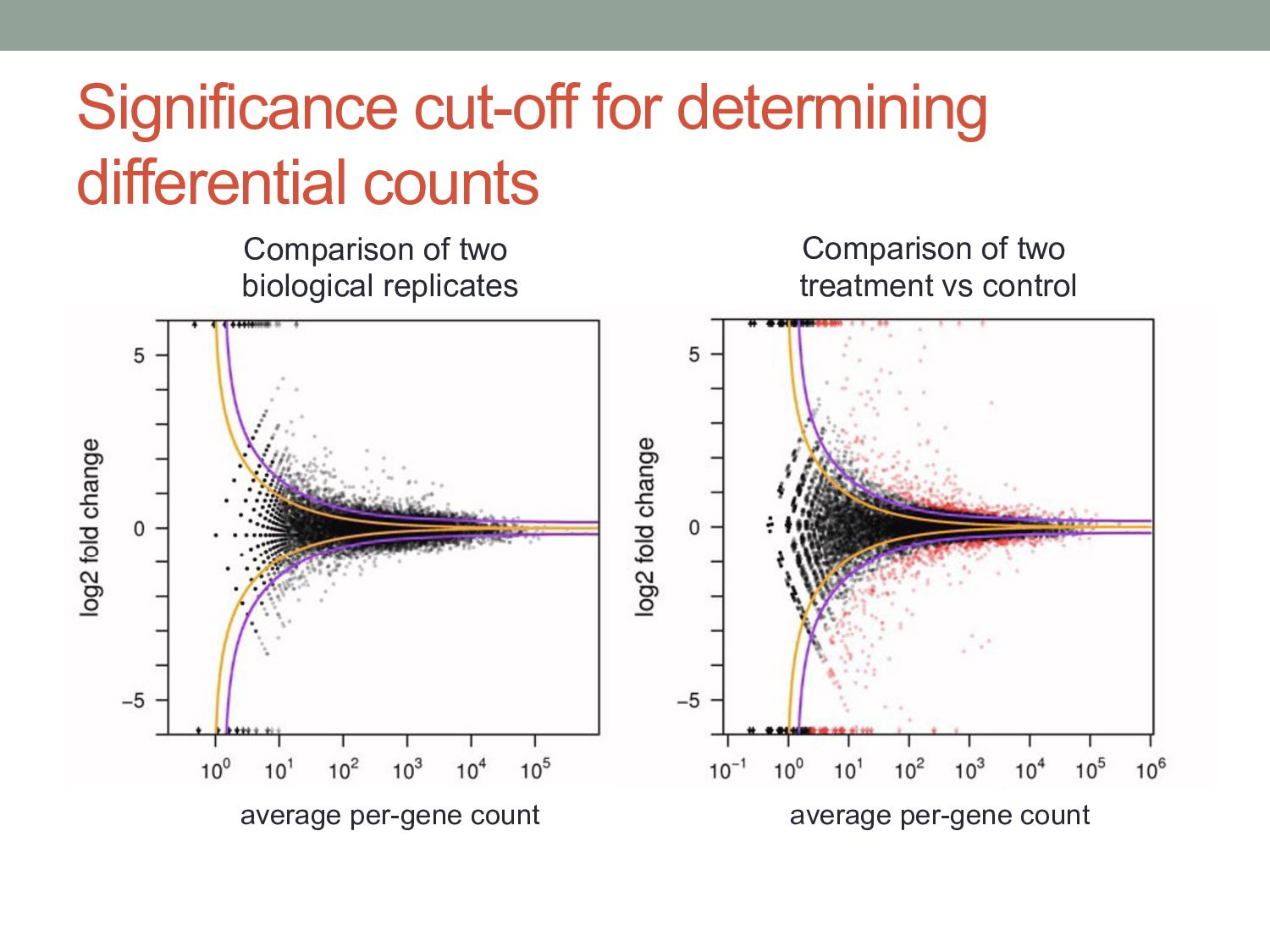
i, test two alternate hypotheses: 1. The level of expression is the same in both conditions. 2. The level of expression is different across conditions. • Likelihood ratio: qi0 defined by combining samples from conditions A & B • Convert to p-value using χ2 distribution. • Correct for multiple hypothesis testing. NB(K iA | S A ,νiA ,q iA )NB(K iB | S B ,νiB ,q iB ) NB(K iA | S A ,νi0 ,q i0 )NB(K iB | S B ,νi0 ,q i0 )
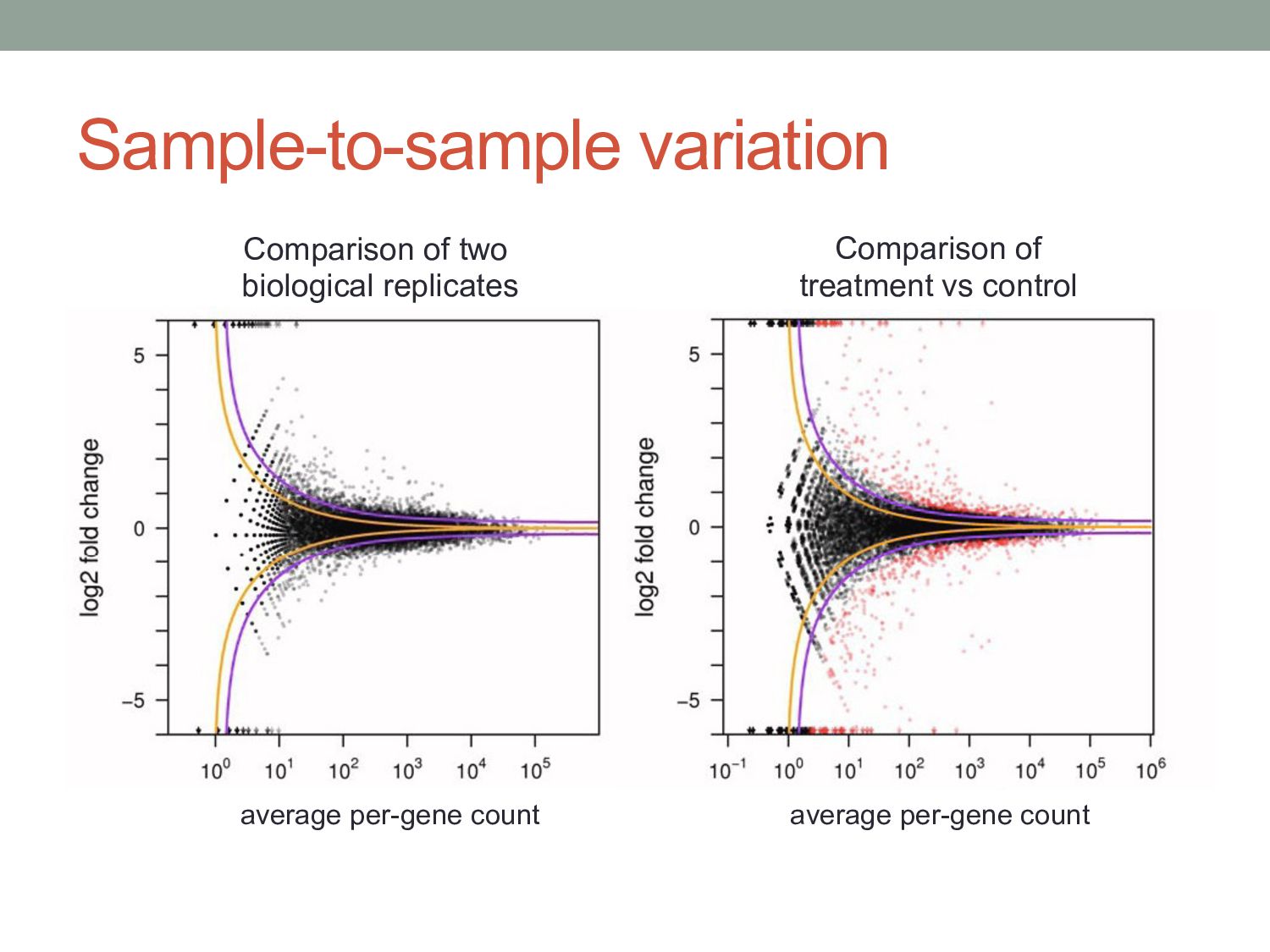
• Analysis task: are ai & bi significantly different? • Observed read count is a noisy measurement of the mean read count expected in a given condition. • The expected mean count depends on the number of mRNA fragments in that condition and the normalization scaling factors.
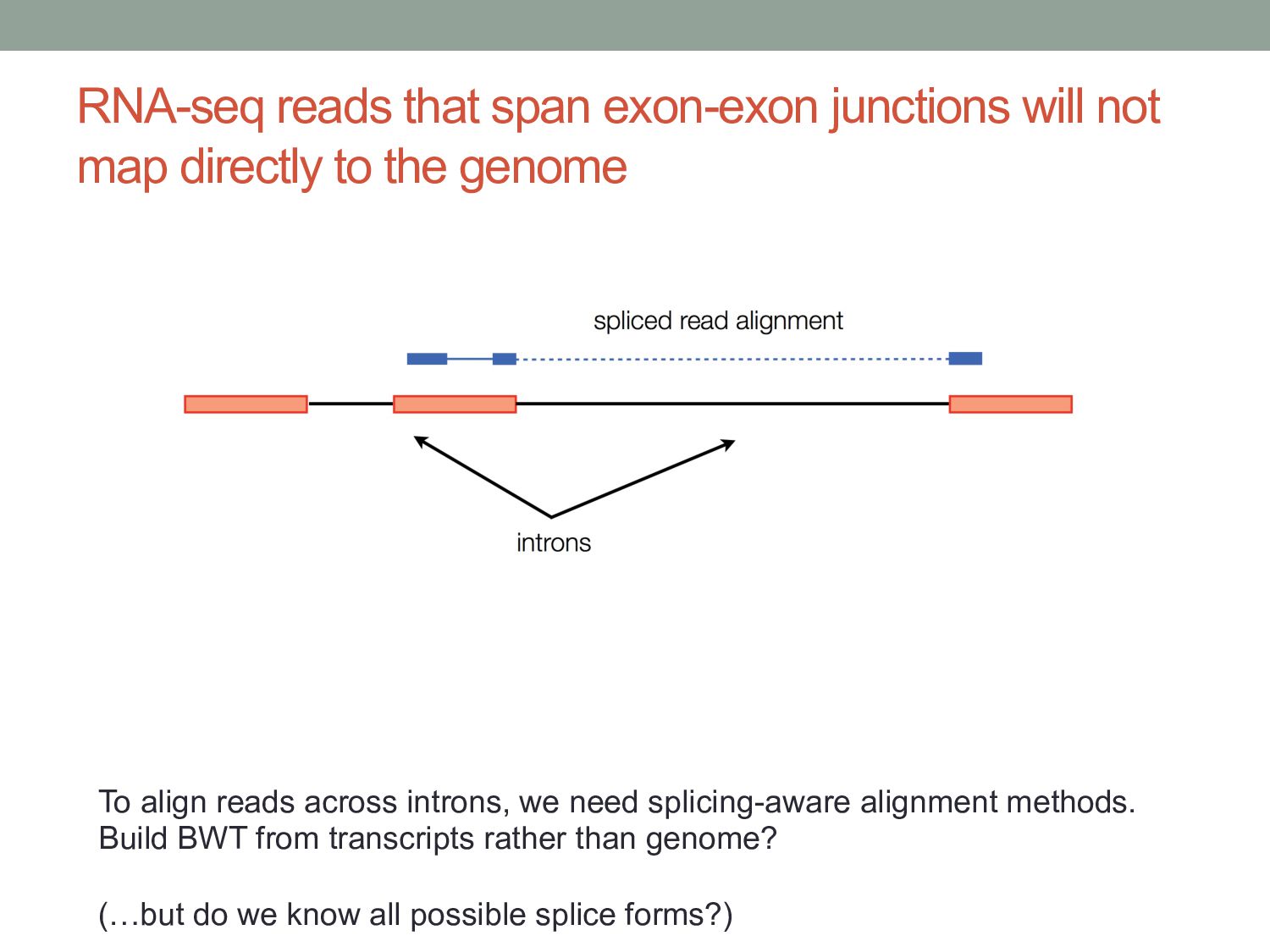
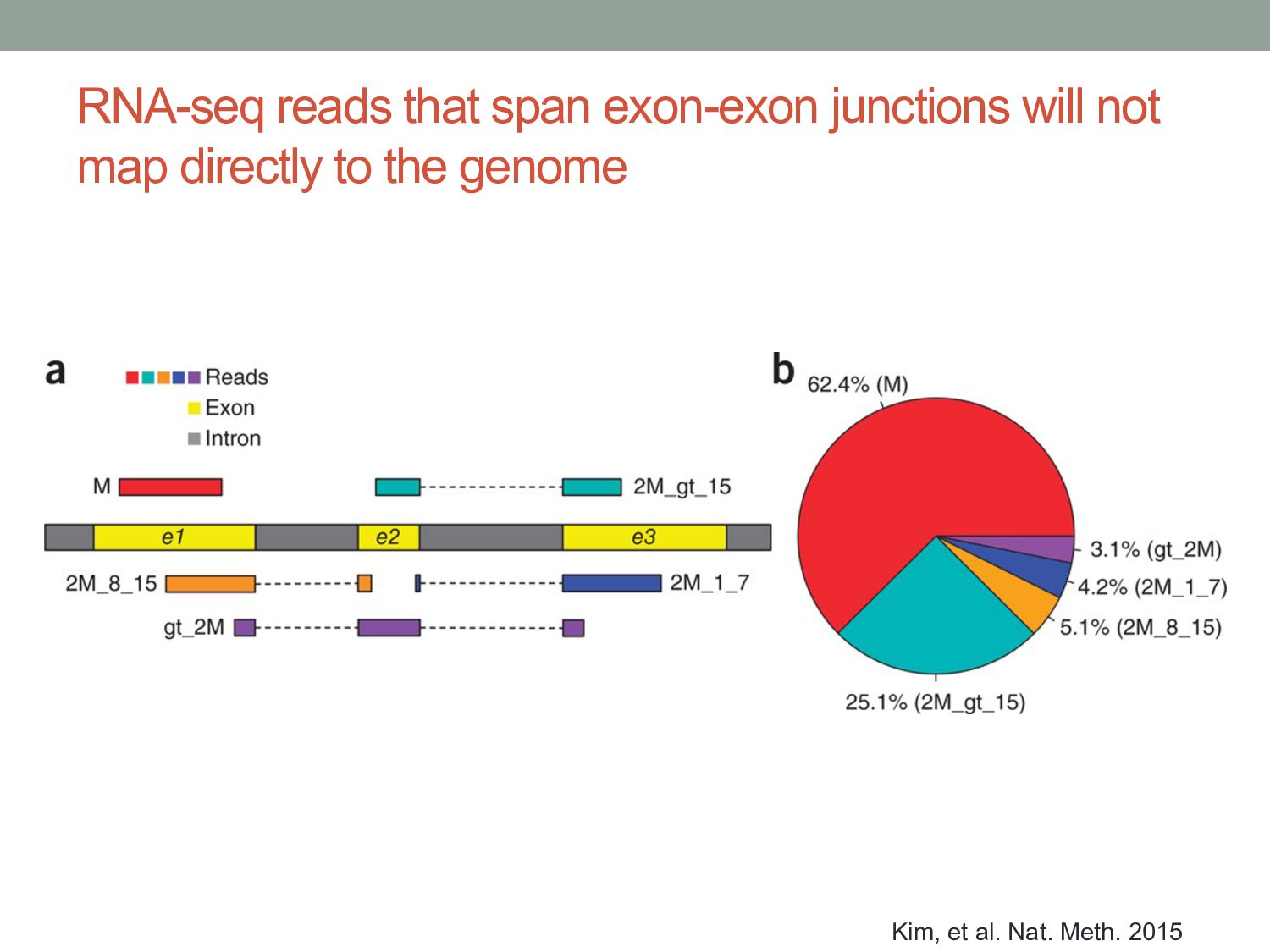
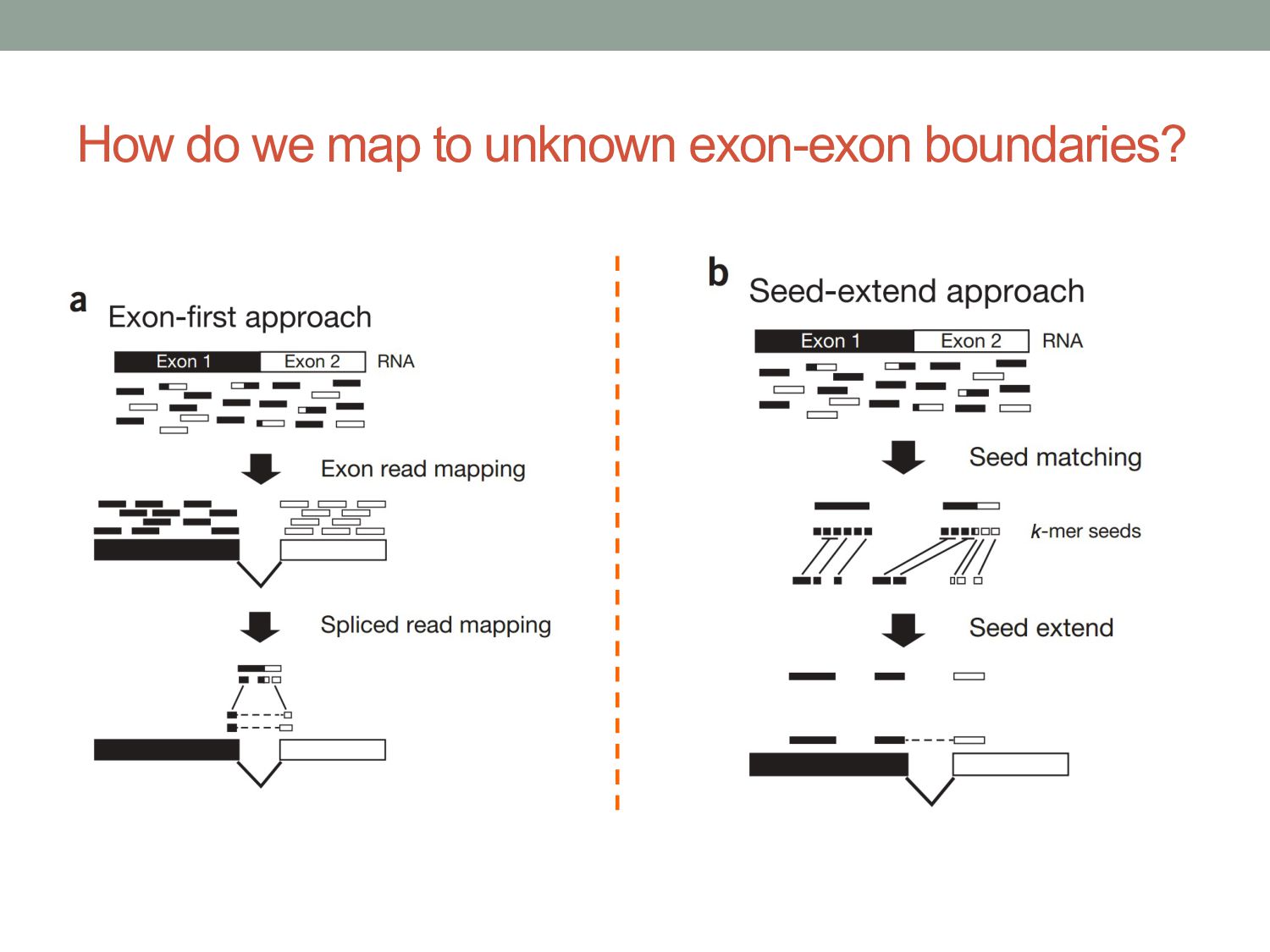
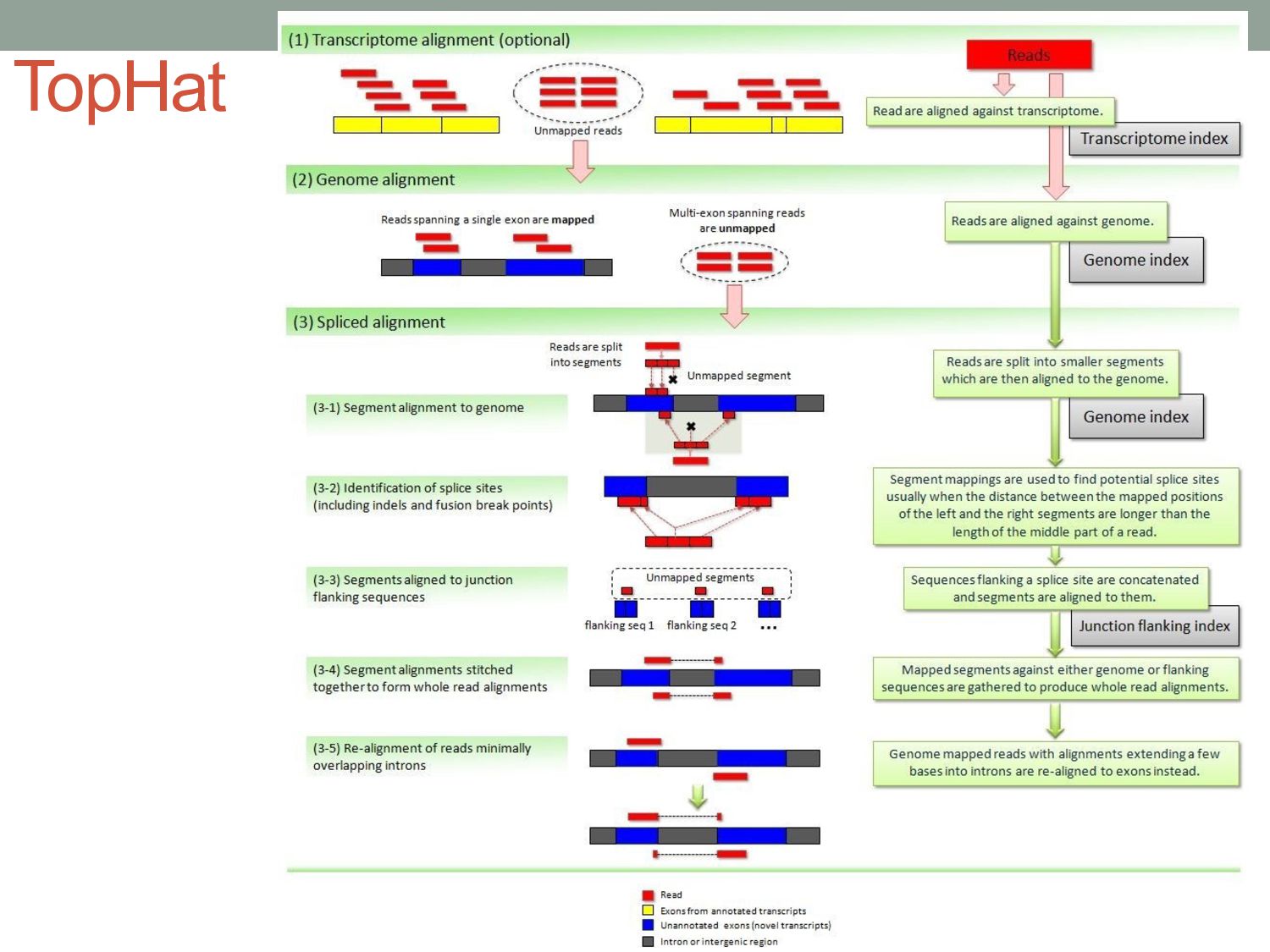
to the genome To align reads across introns, we need splicing-aware alignment methods. Build BWT from transcripts rather than genome? (…but do we know all possible splice forms?)
FM index and a collection of ~55,000 local overlapping 56 kb FM indices. • Finds initial seed locations for potential read alignments using global index and rapidly refines these alignments using corresponding local index. • *(FM index = BWT & some auxiliary data structures) • STAR: • Based on an uncompressed suffix array. • Looks for a maximum mappable prefix (MMP) from the beginning of the read until it can no longer match continuously. • Next tries to find MMP for the unmatched portion of the read
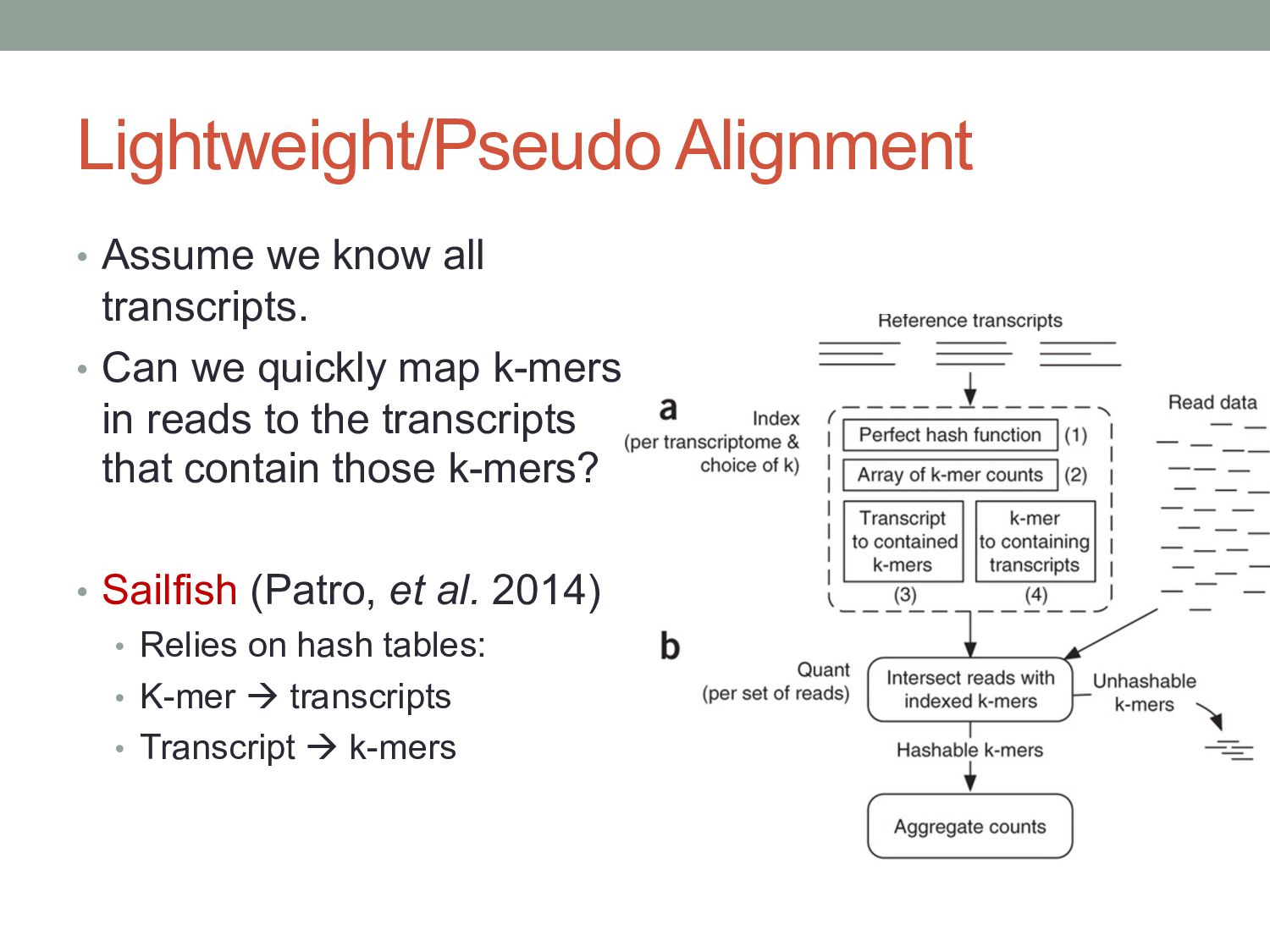
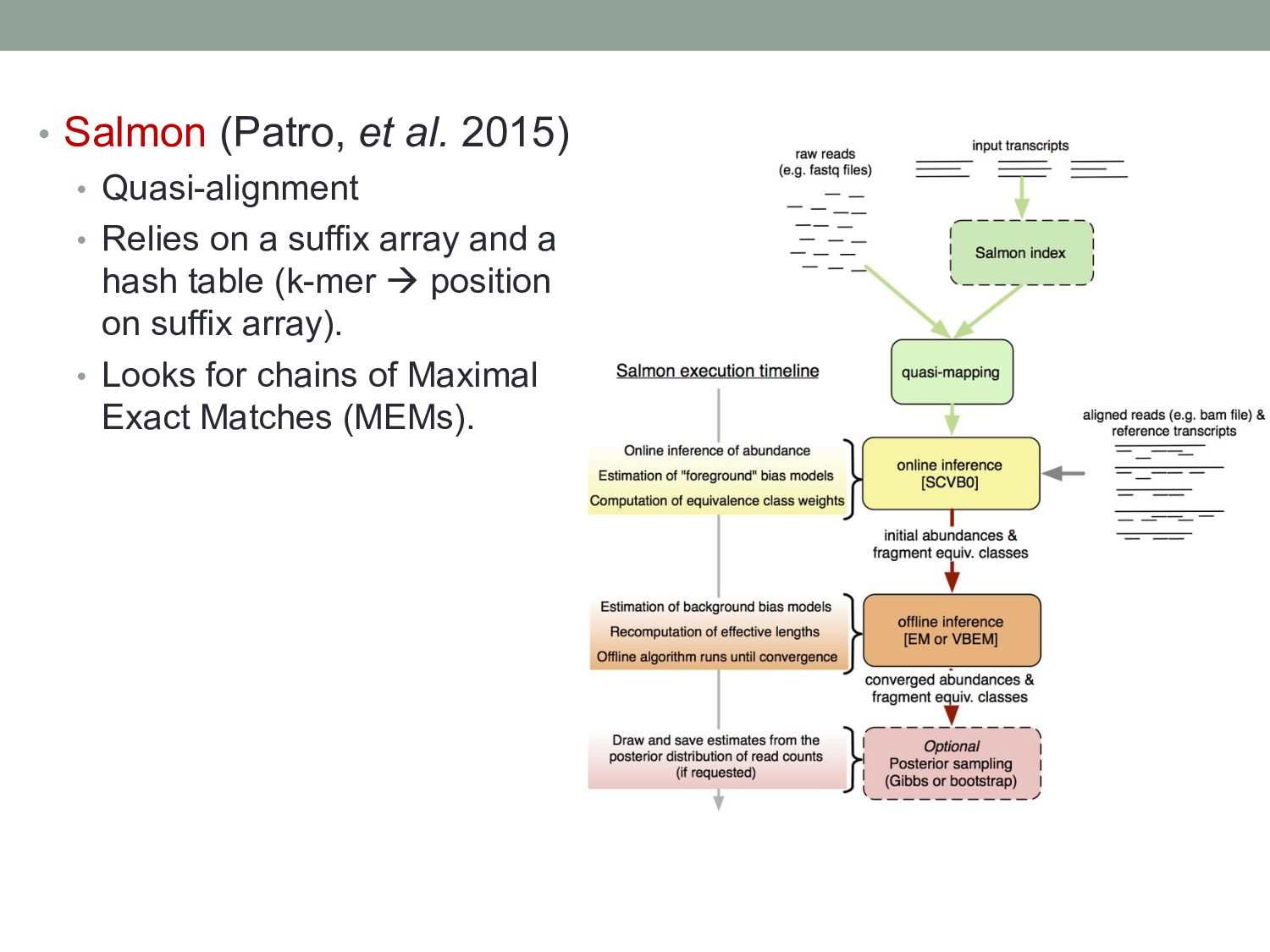
we quickly map k-mers in reads to the transcripts that contain those k-mers? • Sailfish (Patro, et al. 2014) • Relies on hash tables: • K-mer à transcripts • Transcript à k-mers
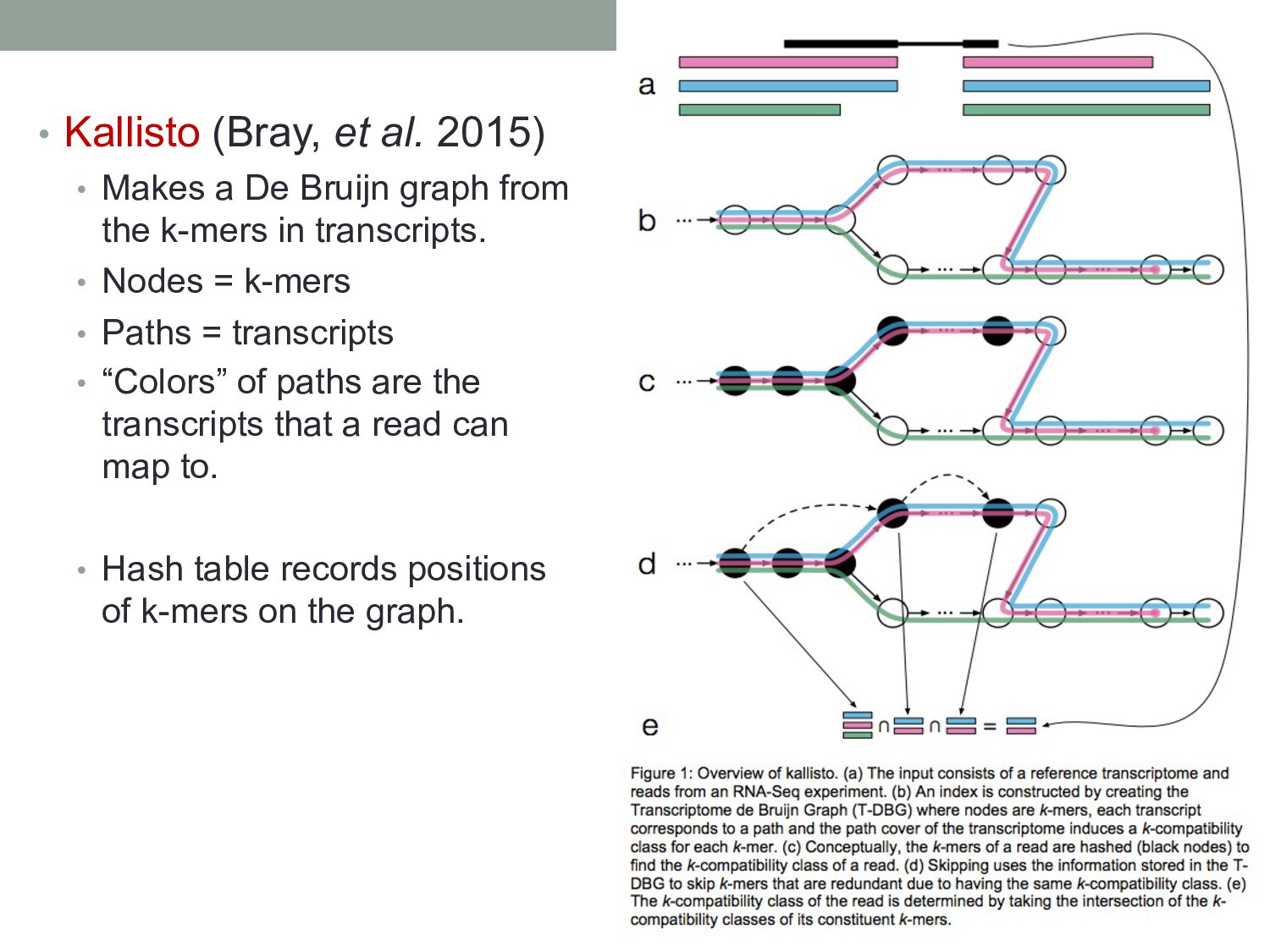
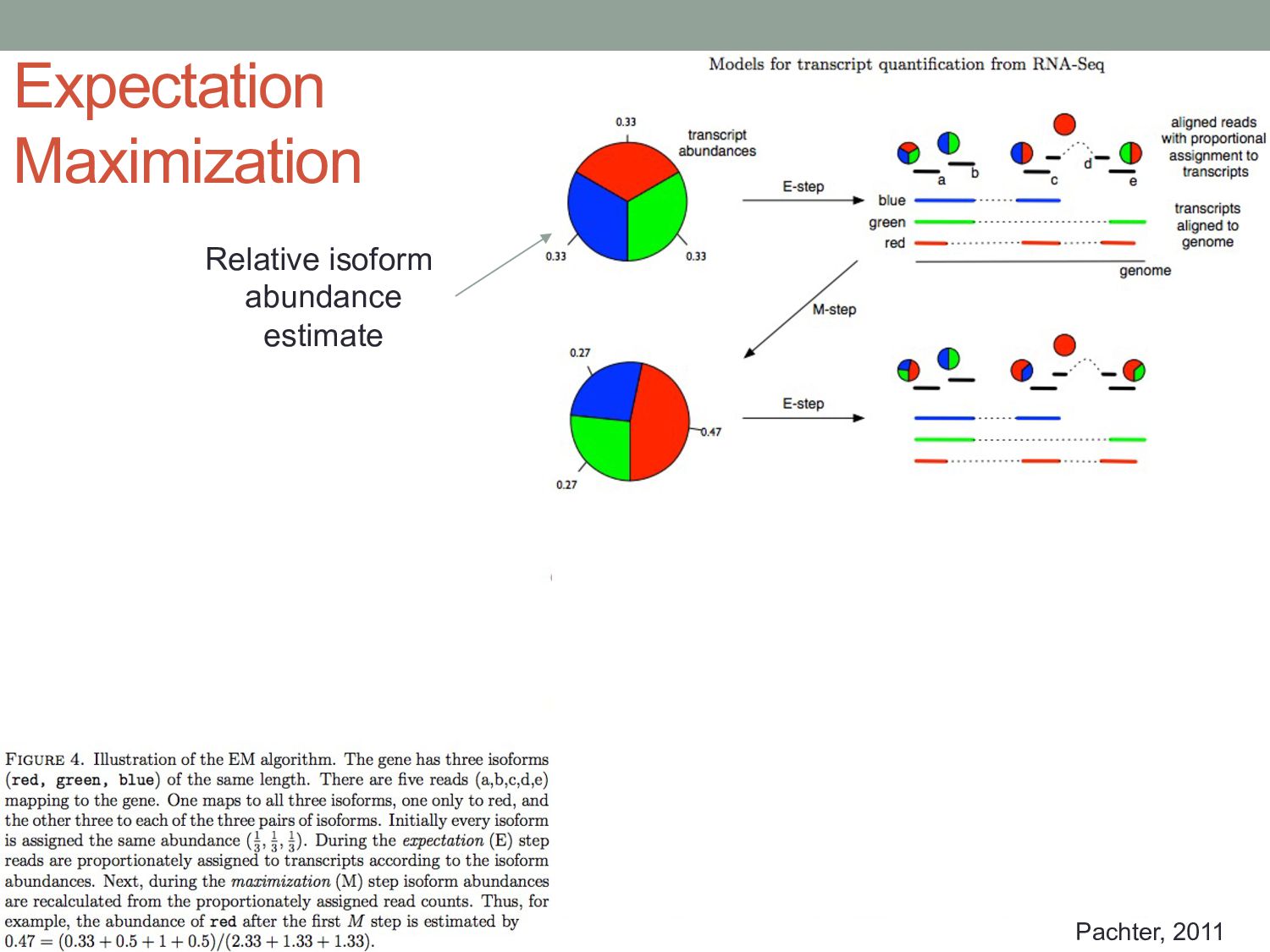
Bruijn graph from the k-mers in transcripts. • Nodes = k-mers • Paths = transcripts • “Colors” of paths are the transcripts that a read can map to. • Hash table records positions of k-mers on the graph.
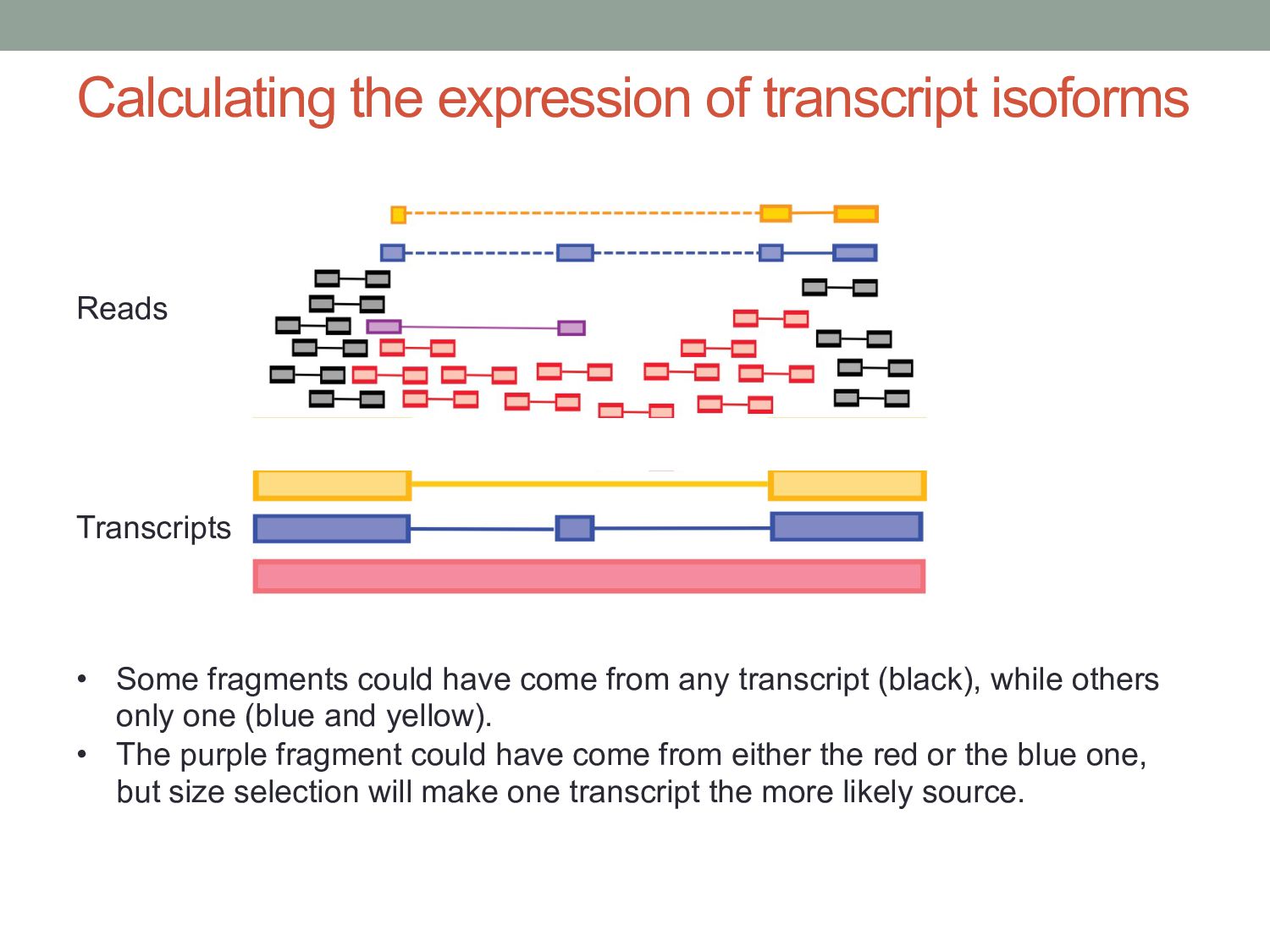
have come from any transcript (black), while others only one (blue and yellow). • The purple fragment could have come from either the red or the blue one, but size selection will make one transcript the more likely source. Reads Transcripts
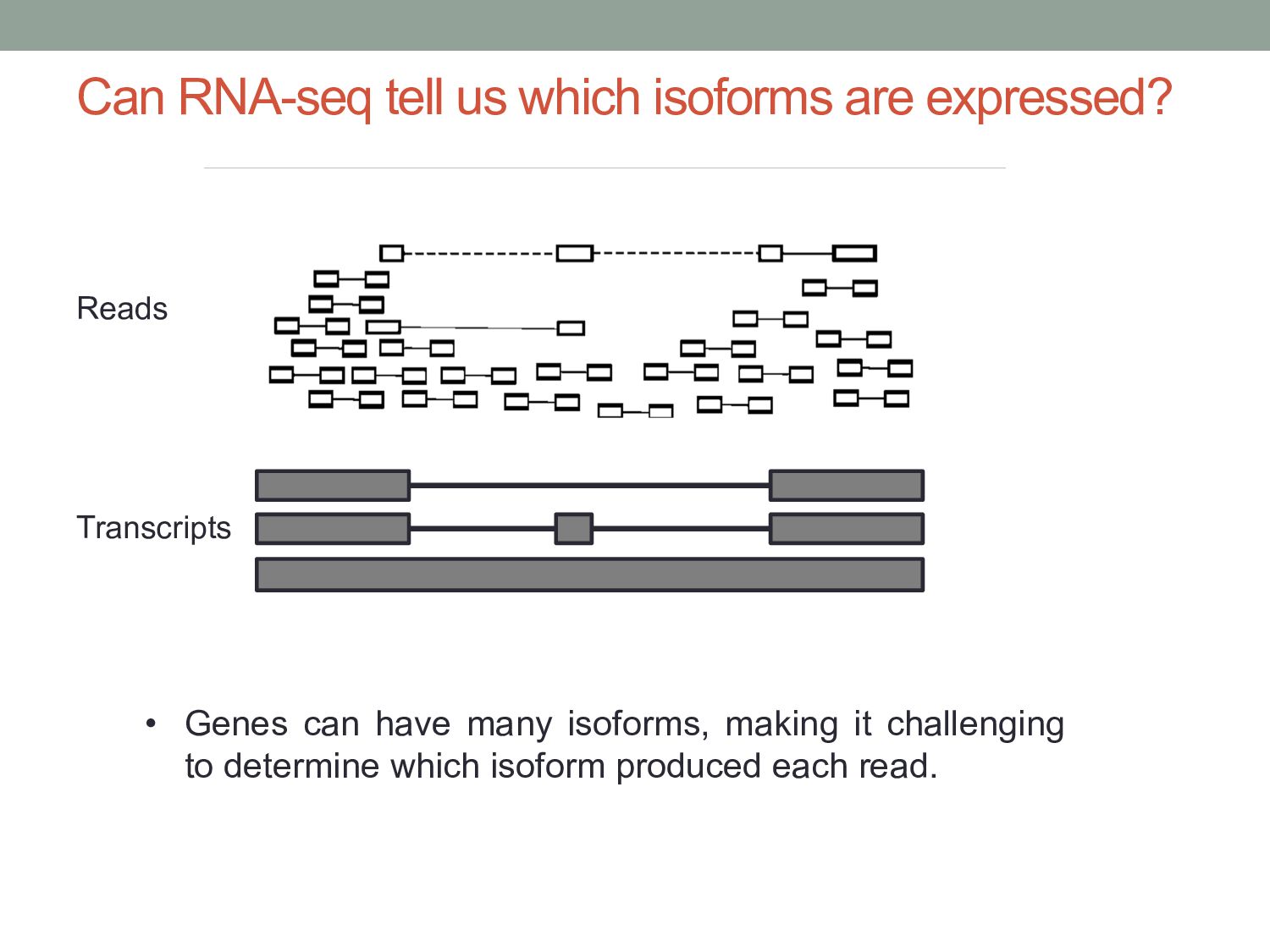
vary across cell types or conditions. With functional genomics assays, this involves analyzing differences in read count information. • Statistical analysis of sequencing (count-based) data needs to account for many sources of variance. • Differentially expressed genes can be found using statistical tests based on the Negative Binomial distribution. • Aligning/quantifying reads to the transcriptome requires methods that are splicing-aware. • Different (pseudo-)alignment methods have trade-offs in speed and sensitivity.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}