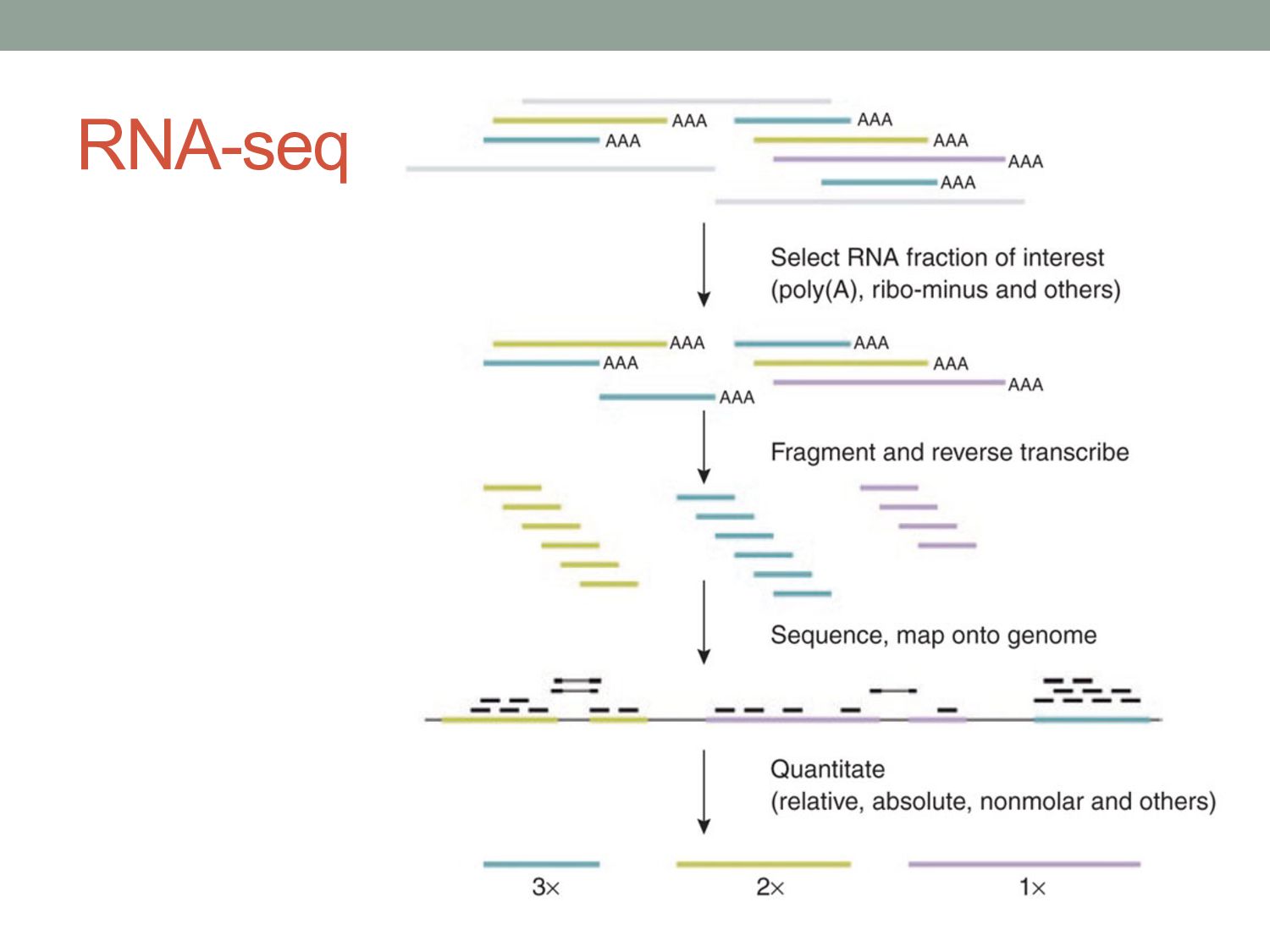
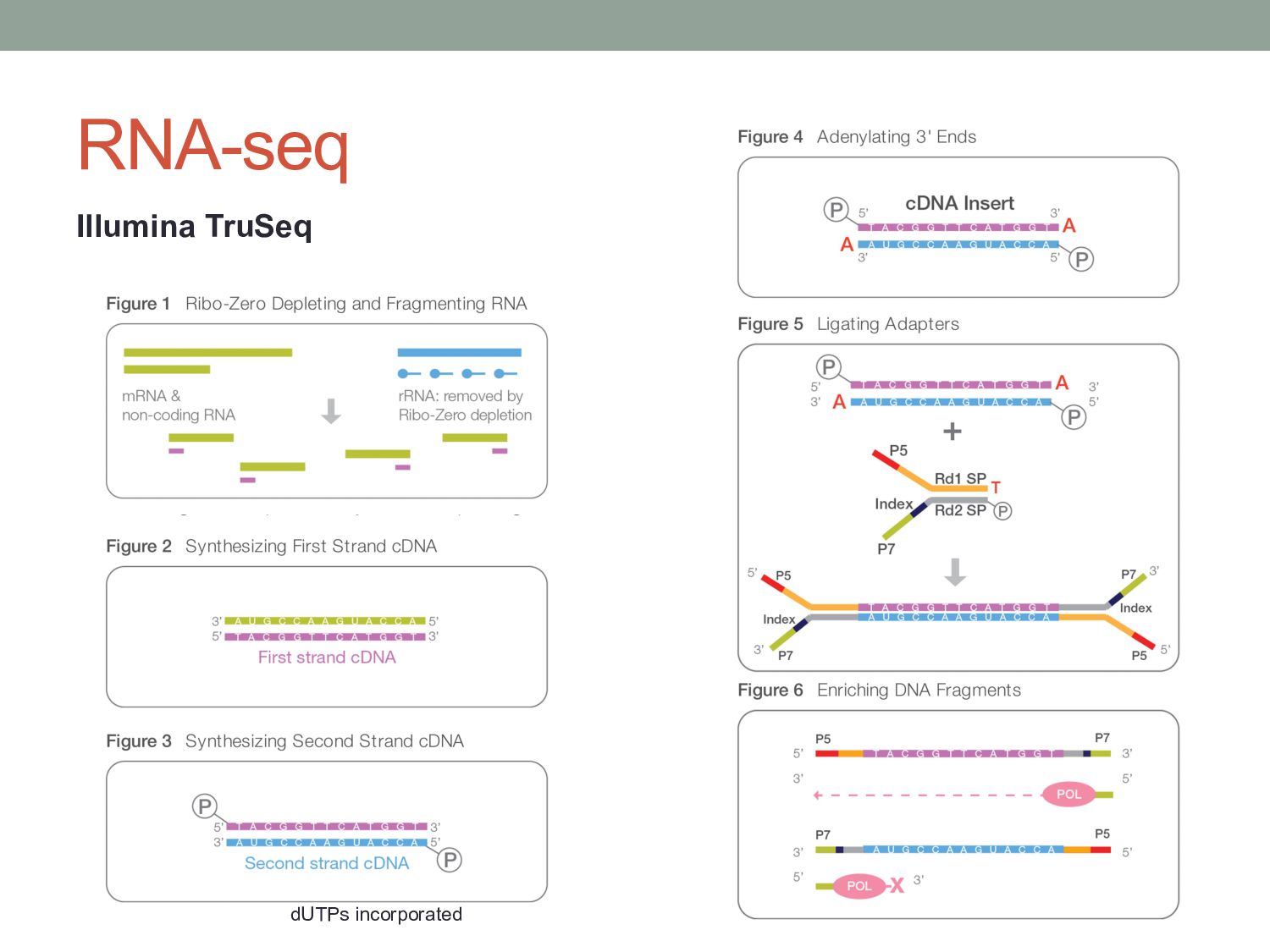
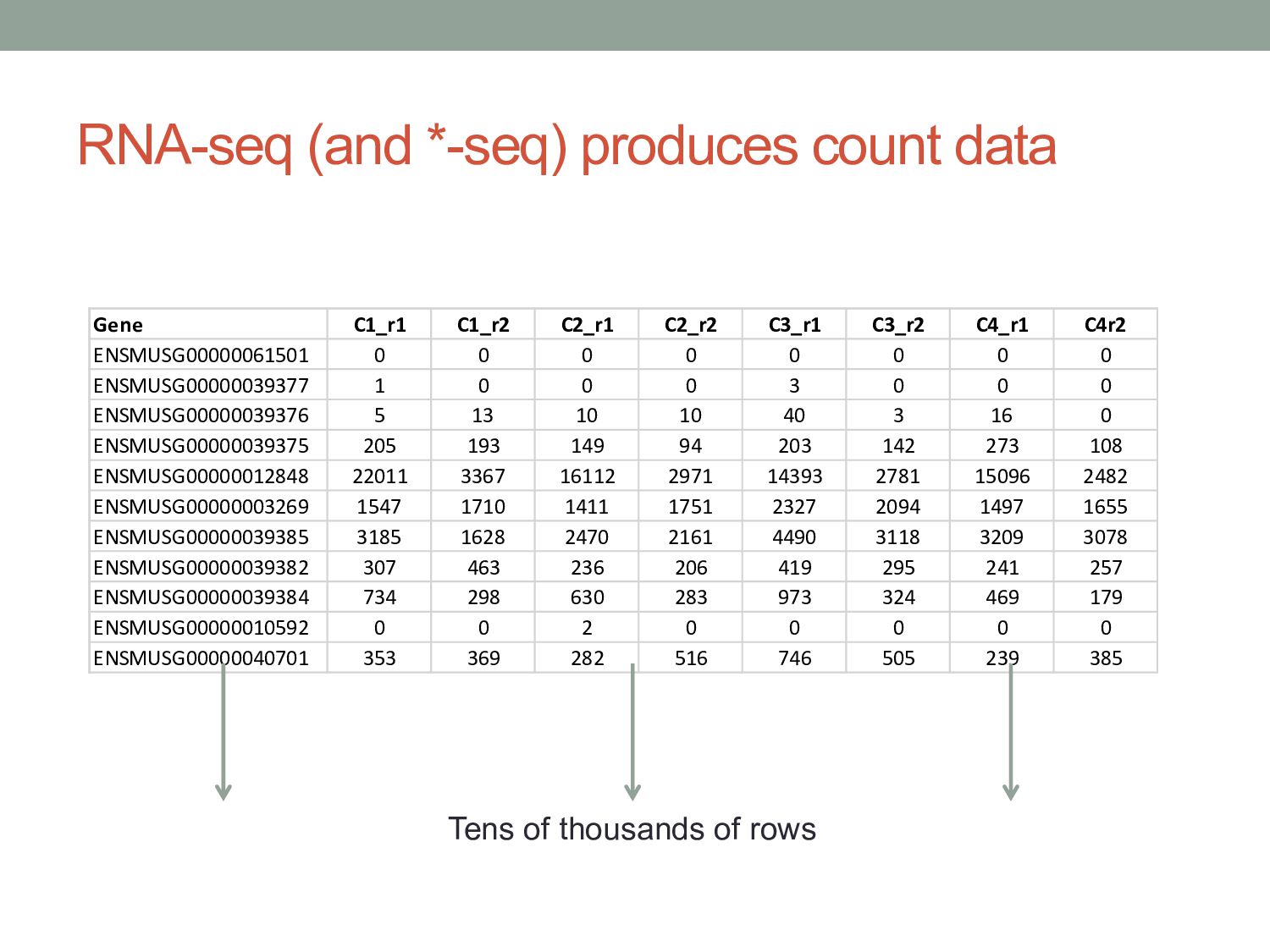
measurements. • Understand statistical approaches to modeling count data from functional genomics experiments. • Discuss other challenges in RNA-seq analysis Some material adapted from: Cole Trapnell Wolfgang Huber
original pool, so count each read at most once. • i.e. length of read doesn’t matter – don’t count nucleotides • Ignore reads if: • Not uniquely aligned to genome • Alignment quality score is bad
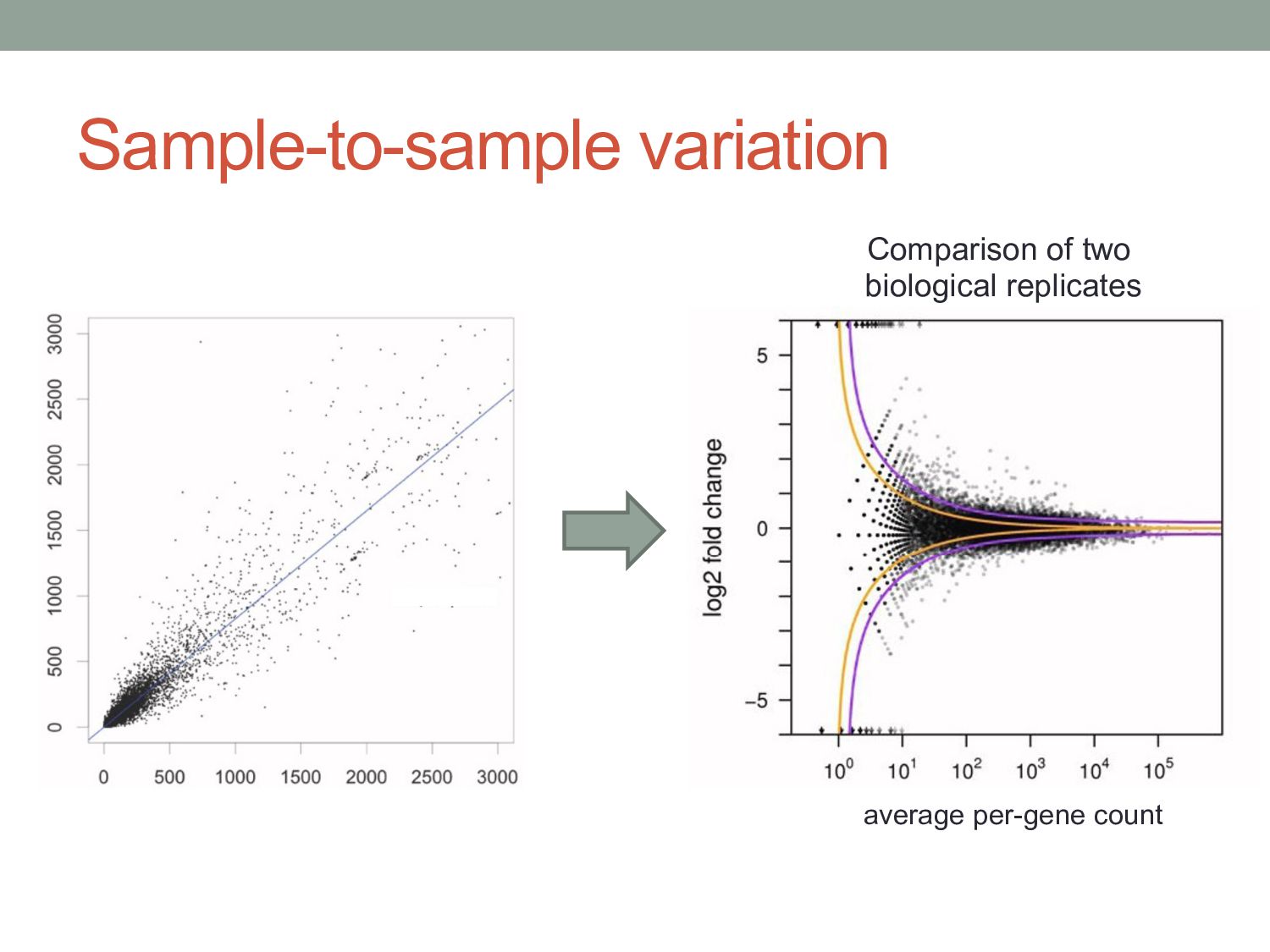
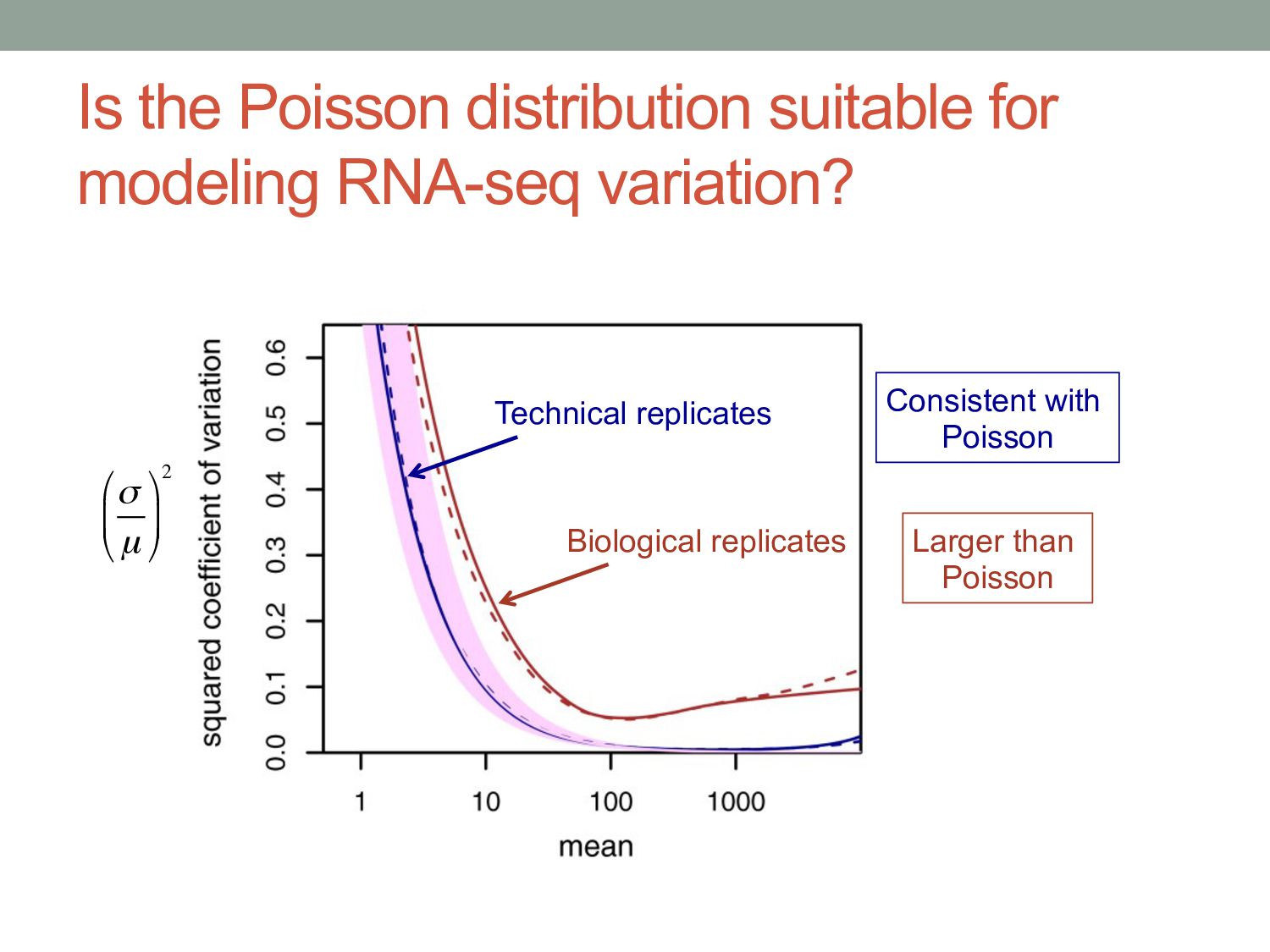
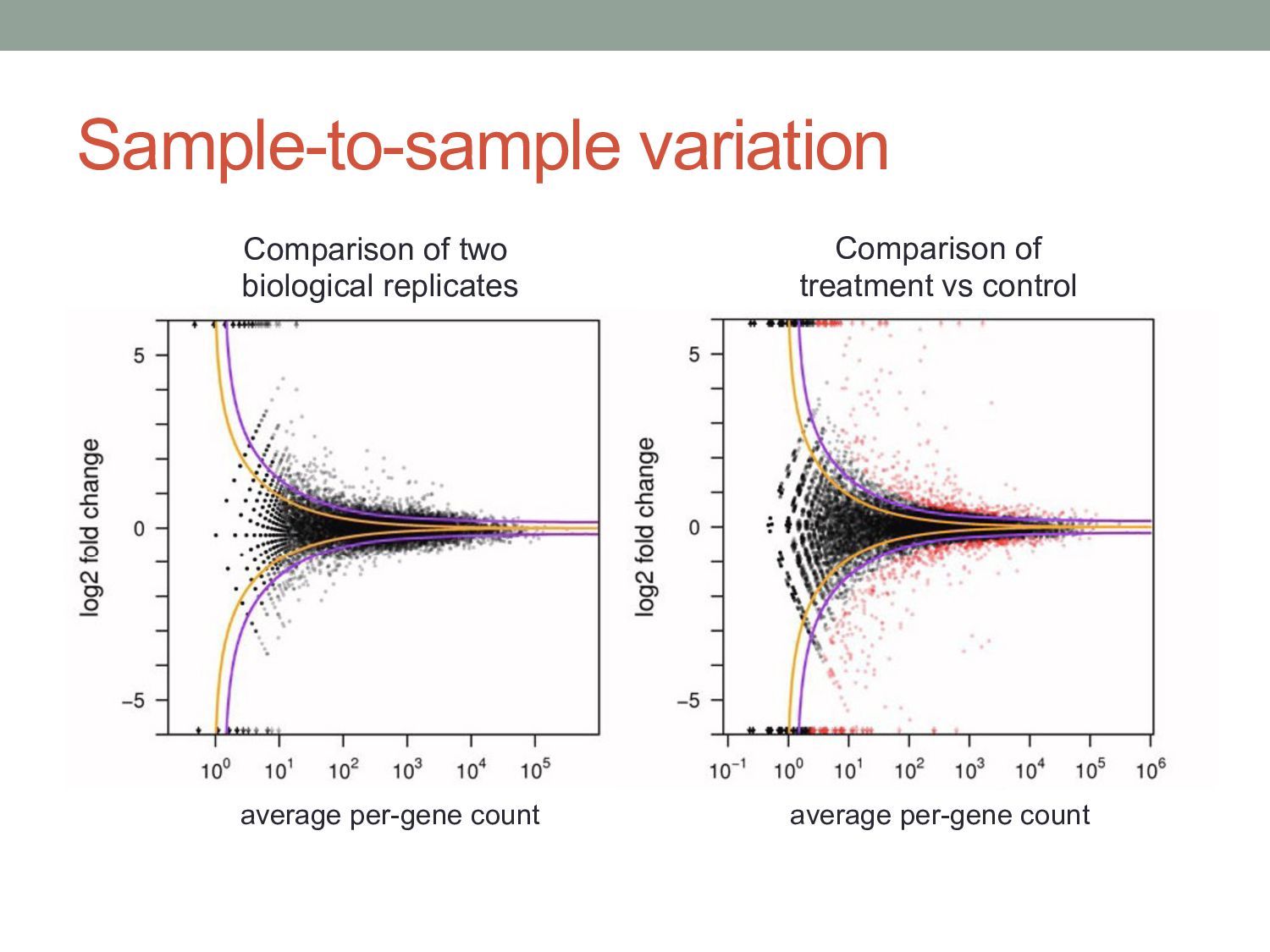
know if the count level for a given gene is significantly different between two samples? • Normalization • Different experiments have different numbers of reads • Different cells may have different levels of total RNA • Read counts are noisy estimates of RNA levels • Repeating the experiment gives different counts for each gene • Need to estimate mean & variance of the counts for each gene • Challenges • Challenge 1: Normal distribution doesn’t work very well for count data • Challenge 2: Typically few replicates for RNA-seq / ChIP-seq • Challenge 3: Variance different for low counts vs. high counts
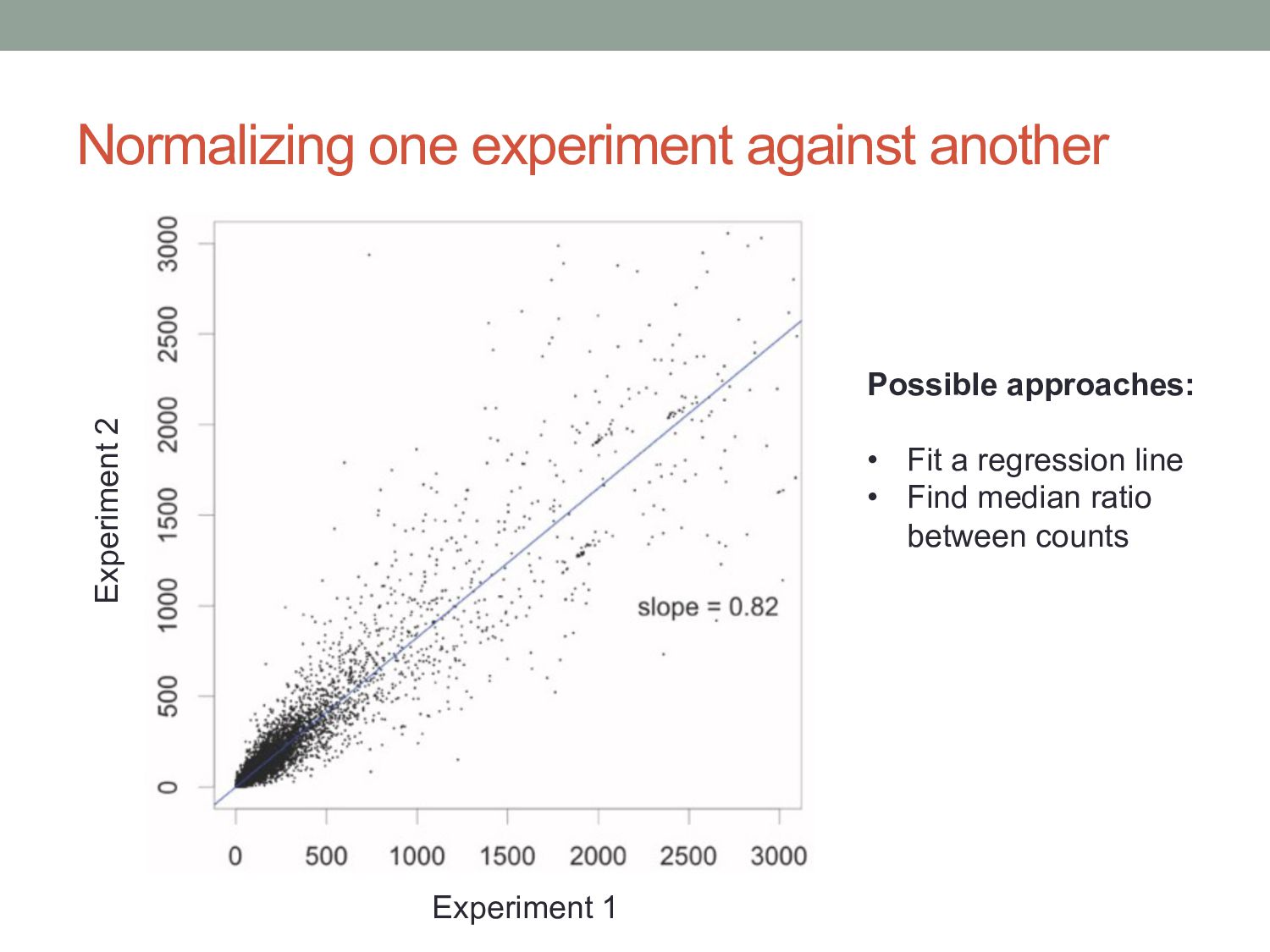
deeper than sample B, we expect the counts of genes to be higher in A than B. • Should we just divide through by the total number of reads? A B Gene 1 10 18 Gene 2 30 58 Gene 3 20 42 Gene 4 40 82 Total reads 100 200 A B Gene 1 20 10 Gene 2 30 15 Gene 3 40 20 Gene 4 10 55 Total reads 100 100
• Analysis task: are ai & bi significantly different? (or: reject hypothesis that the are expressed at same level) • Observed read count is a noisy measurement of the mean read count expected in a given condition. • The expected mean count depends on the number of mRNA fragments in that condition and the normalization scaling factors.
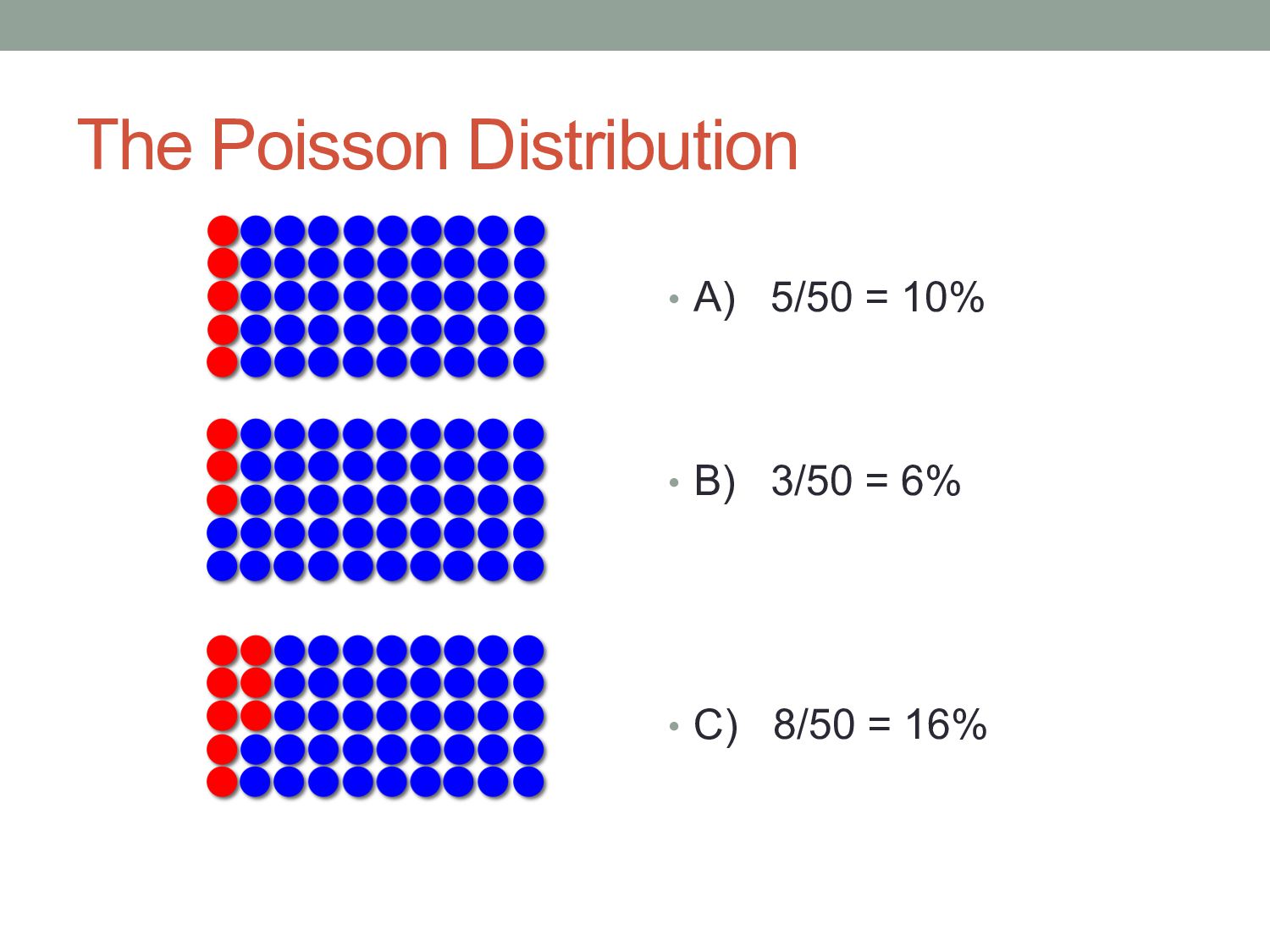
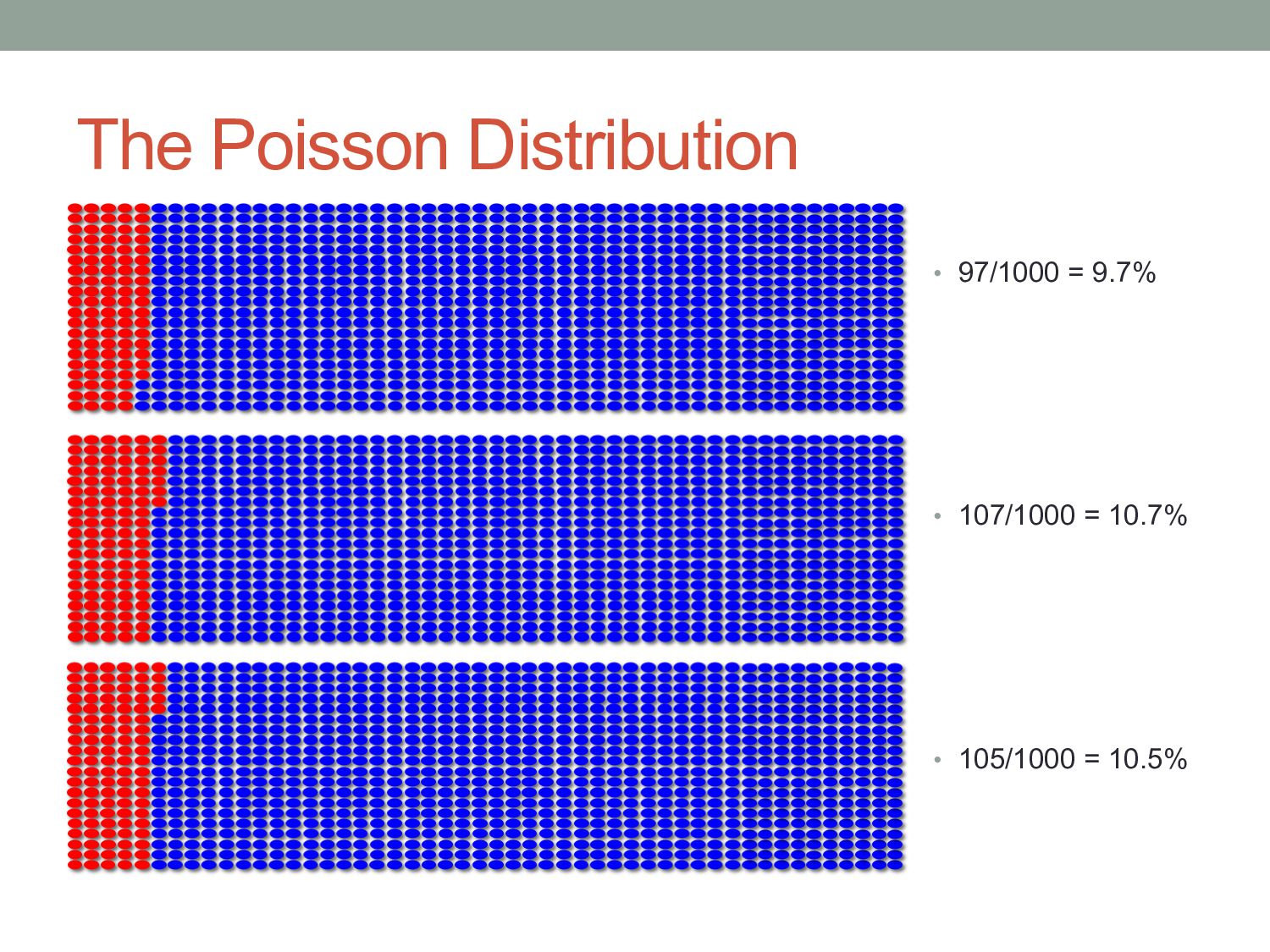
of which are red. • Several volunteers are asked to estimate the percentage of beads. • Each is permitted to draw 50 beads from the bag. The action of drawing red beads from the bag is an example of Poisson sampling – the number of red beads drawn after a number of samples follows a Poisson distribution.
variance Expected number of red beads Standard deviation of number of red beads Relative standard deviation 10 100 1,000 10,000 √10 = 3.16 √100 = 10.0 √1,000 = 31.6 √10,000 = 100.0 31.6% 10.0% 3.16% 1.0%
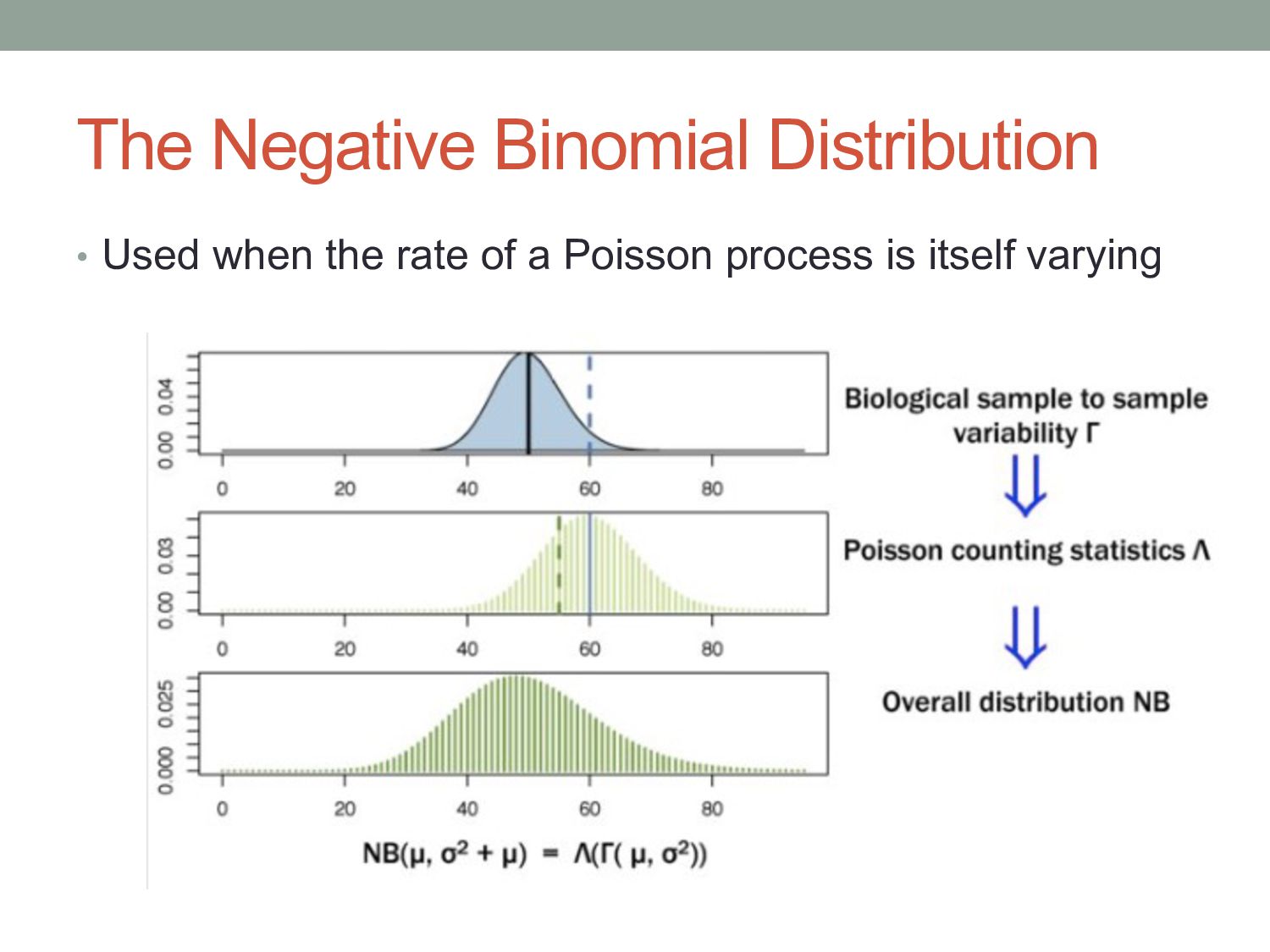
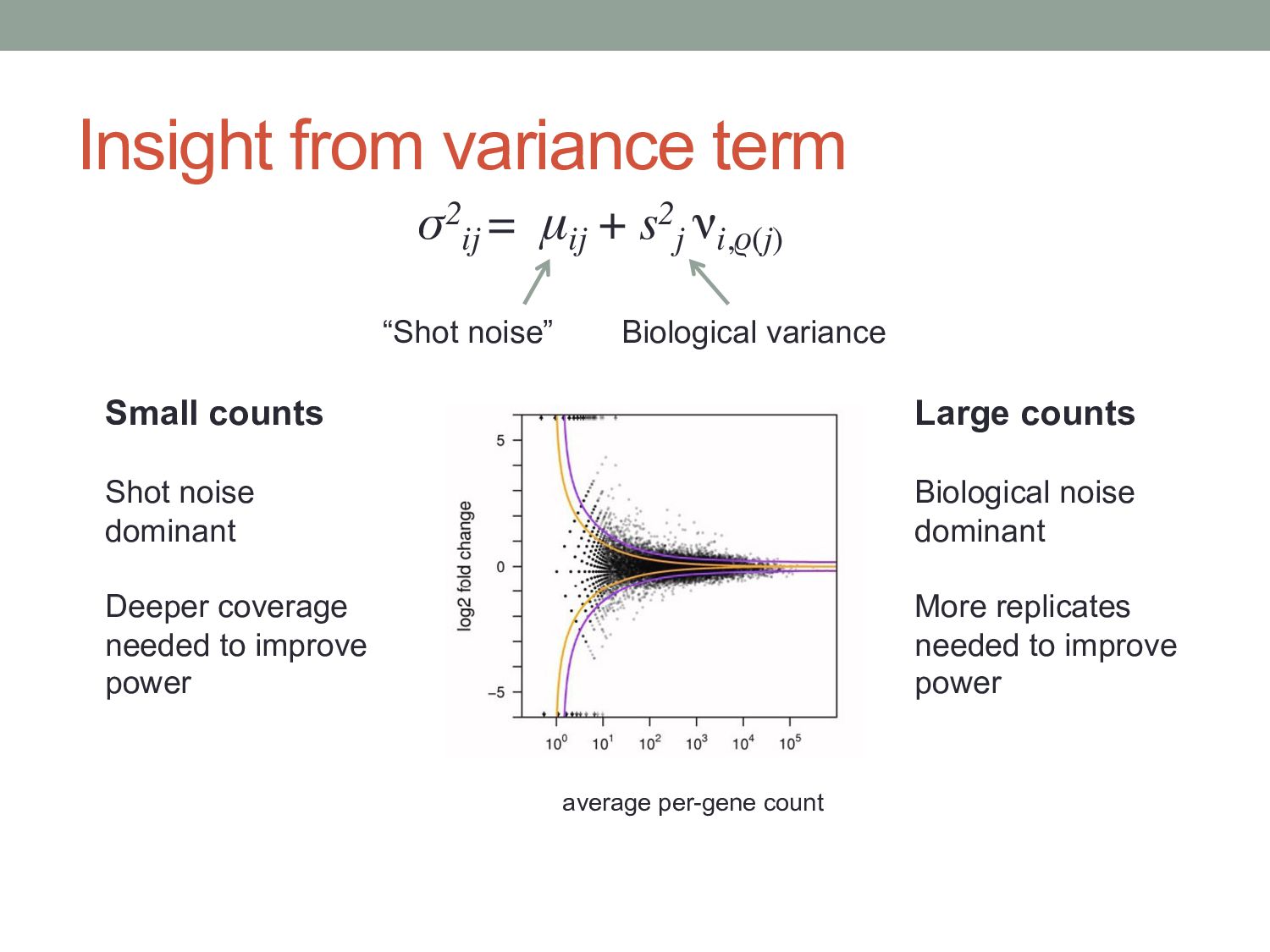
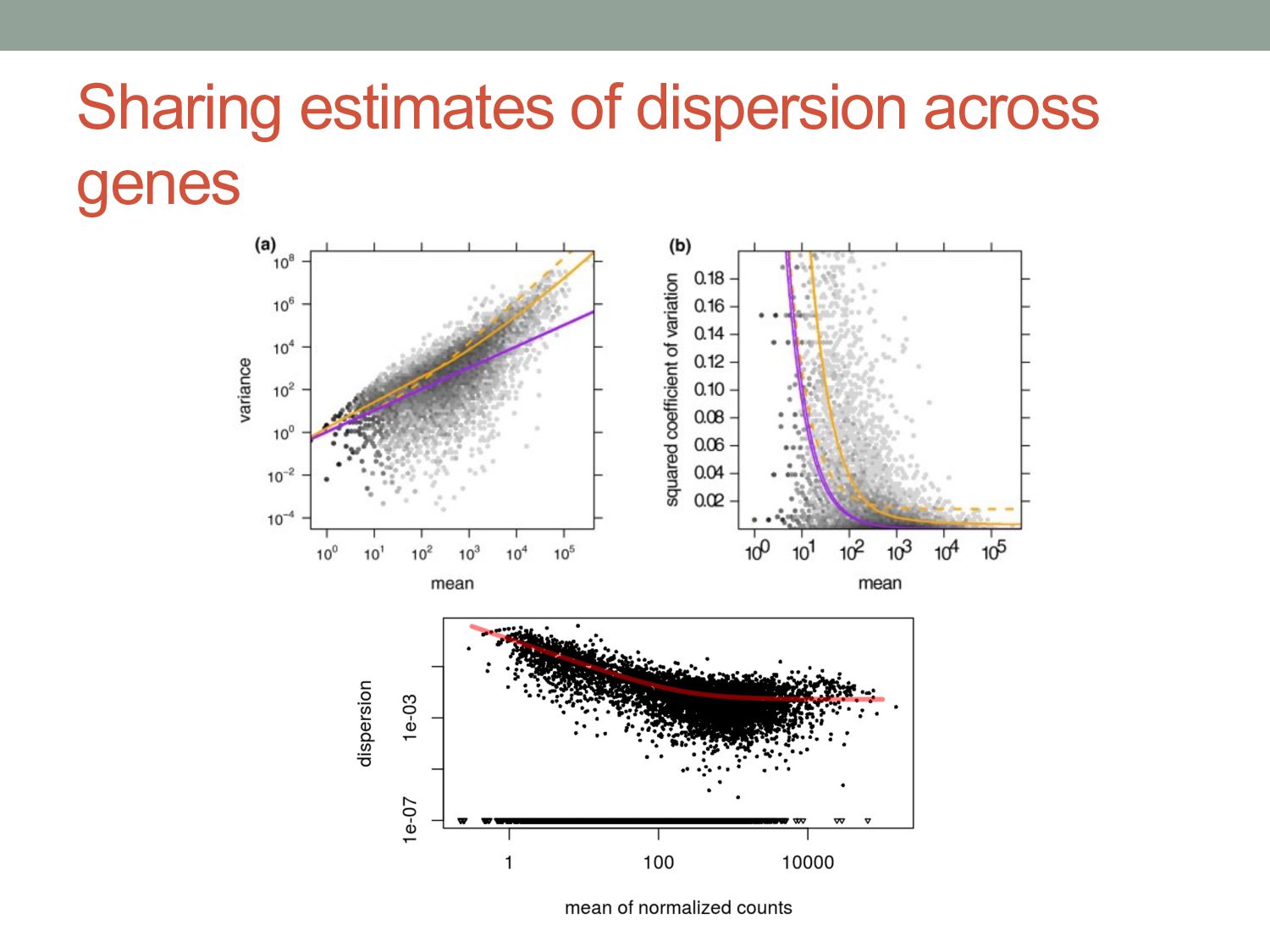
i in conditionx depends on a Negative Binomial (NB) function. • The mean of the NB depends on the concentration of mRNA fragments for gene i in condition x. • The variance of the NB is a combination of two effects: • “Shot noise”; equal to the mean of the NB – i.e. the Poisson effect. + • Biological variance between replicates. • We estimate the NB variance parameter by observing how variable the counts are across biological replicates.
) i = gene j = sample kij = count for gene i in sample j qi,ρ(j) = expected concentration of fragments of gene i in condition ρ(j) sj = scaling factor νi,ρ(j) = per gene variance estimated from smooth function of q Where: μij =qi,ρ(j) sj σ2 ij = μij + s2 j νi,ρ(j) “Shot noise” Biological variance between replicates mean: variance:
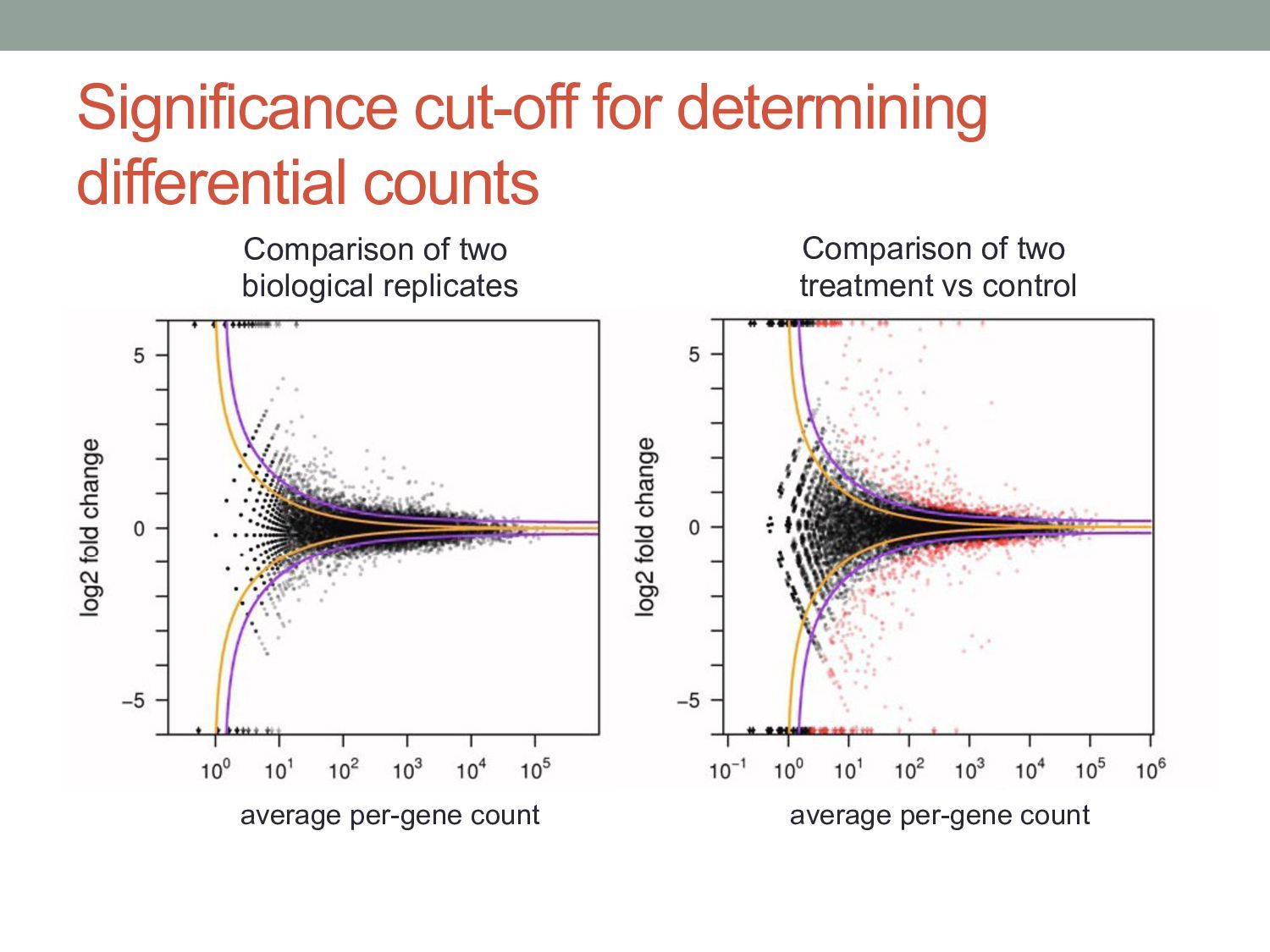
μij + s2 j νi,ρ(j) “Shot noise” Biological variance Small counts Shot noise dominant Deeper coverage needed to improve power Large counts Biological noise dominant More replicates needed to improve power
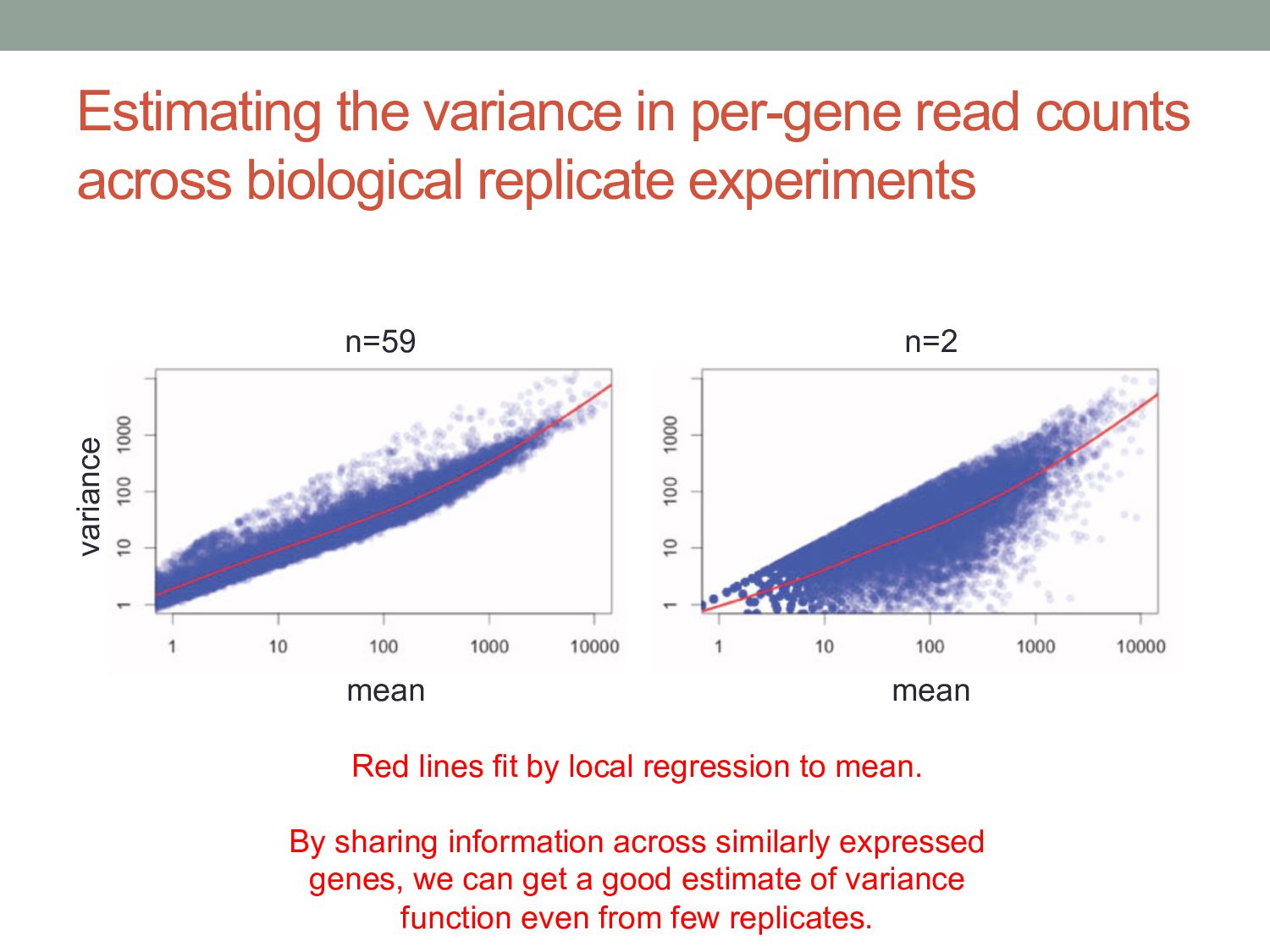
experiments n=59 n=2 mean mean variance Red lines fit by local regression to mean. By sharing information across similarly expressed genes, we can get a good estimate of variance function even from few replicates.
i, test two alternate hypotheses: 1. The level of expression is the same in both conditions. 2. The level of expression is different across conditions. • Likelihood ratio: qi0 defined by combining samples from conditions A & B • Convert to p-value using χ2 distribution. • Correct for multiple hypothesis testing. NB(K iA | S A ,νiA ,q iA )NB(K iB | S B ,νiB ,q iB ) NB(K iA | S A ,νi0 ,q i0 )NB(K iB | S B ,νi0 ,q i0 )
• Analysis task: are ai & bi significantly different? • Observed read count is a noisy measurement of the mean read count expected in a given condition. • The expected mean count depends on the number of mRNA fragments in that condition and the normalization scaling factors.
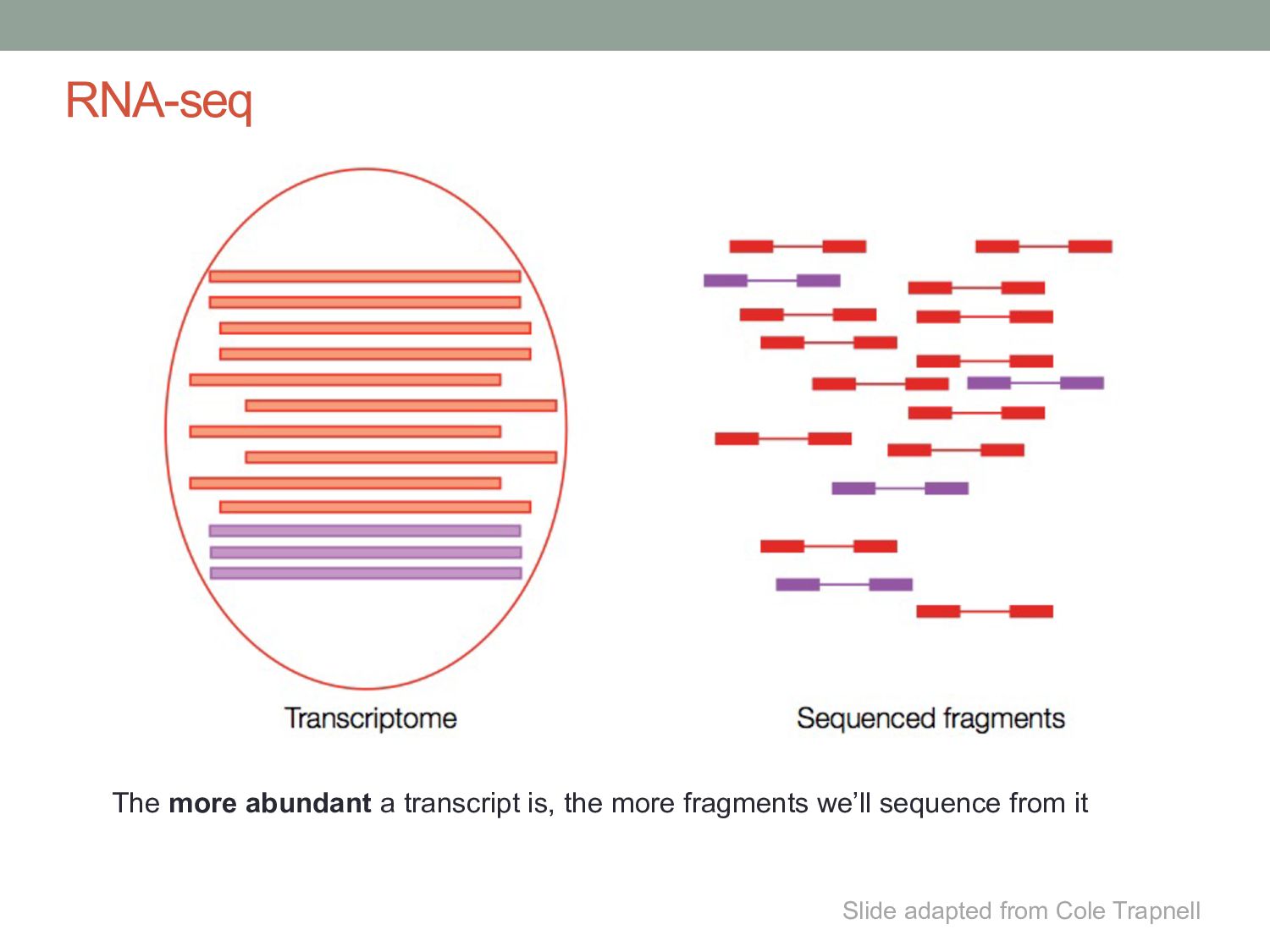
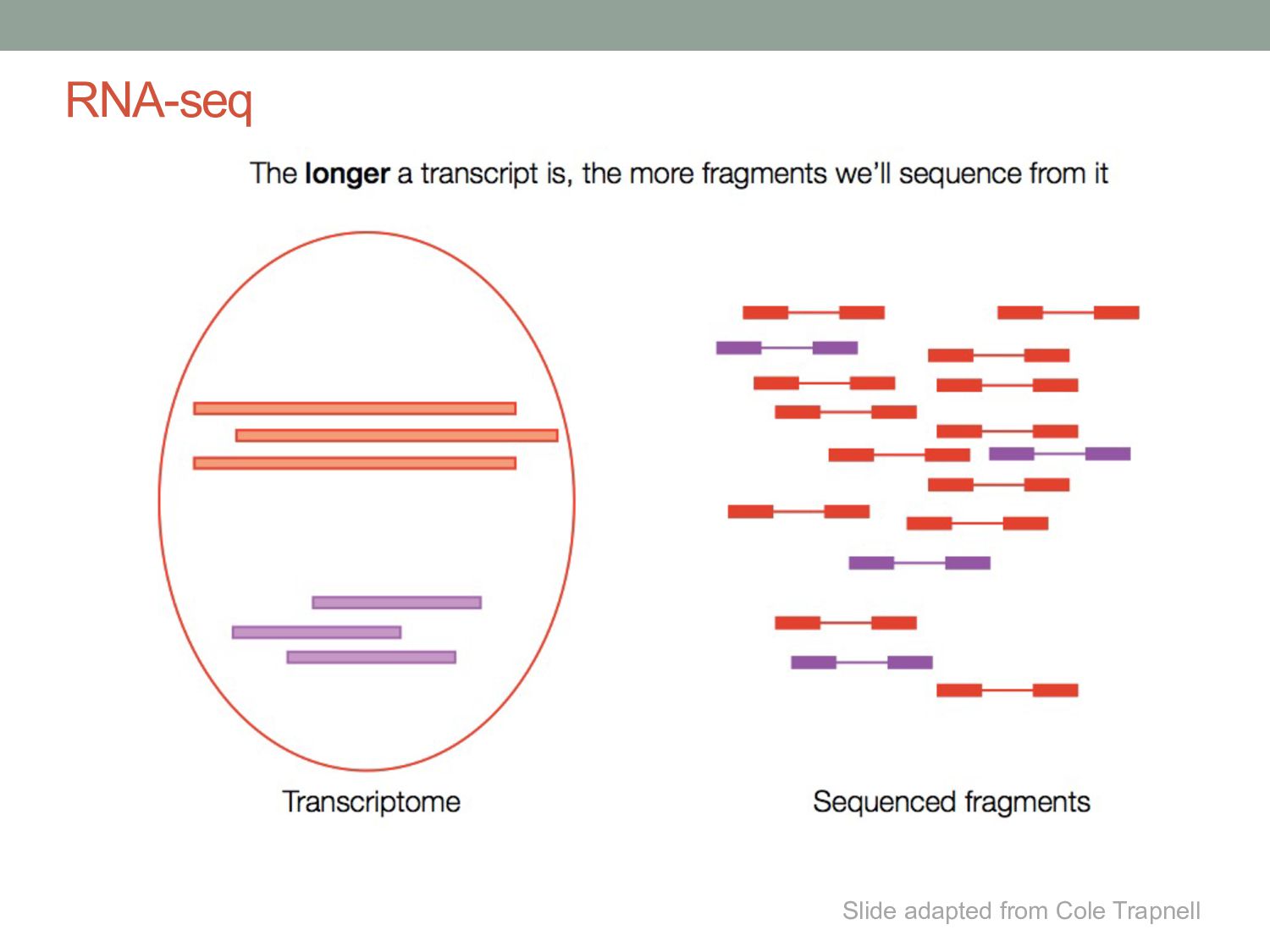
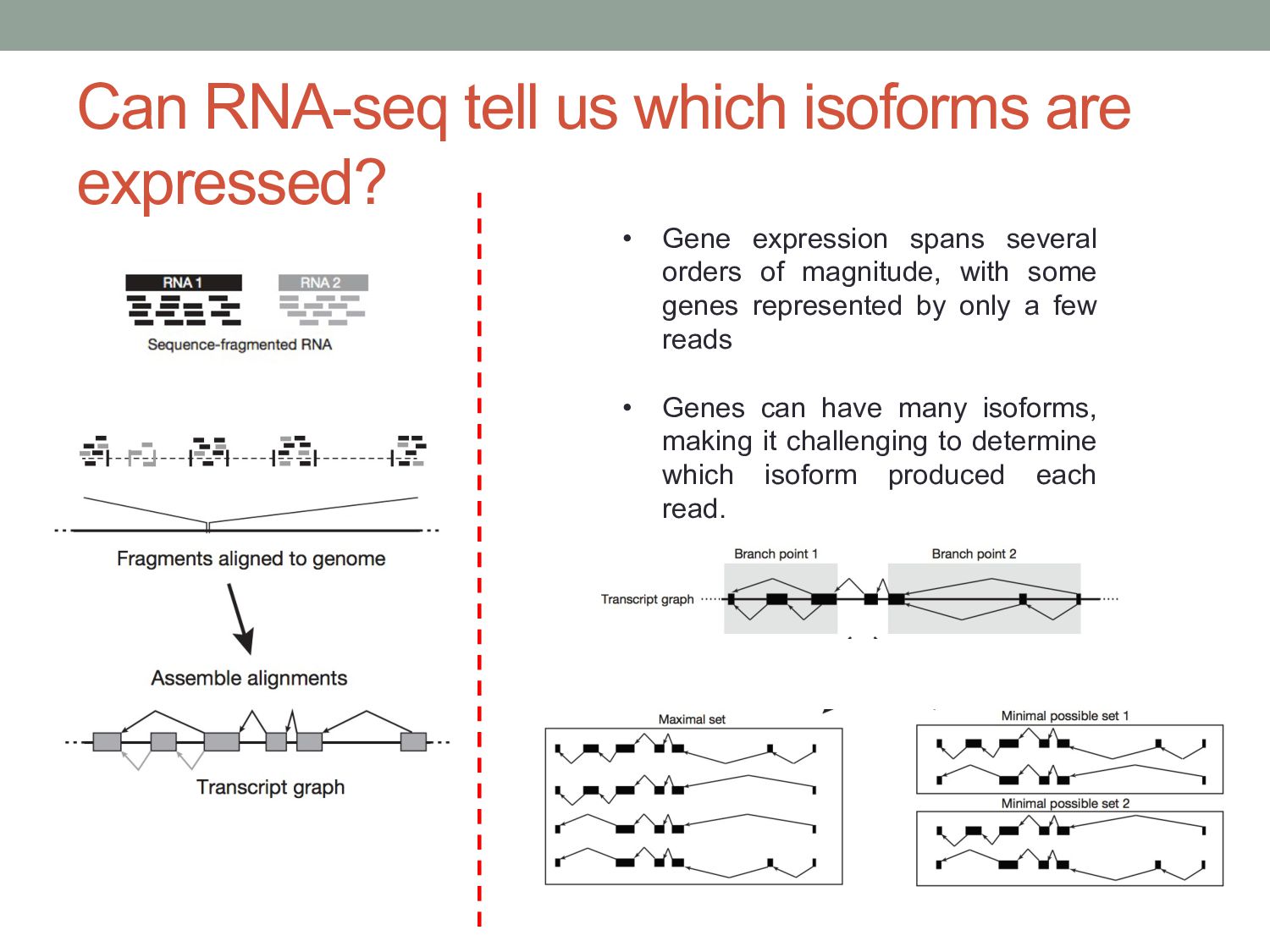
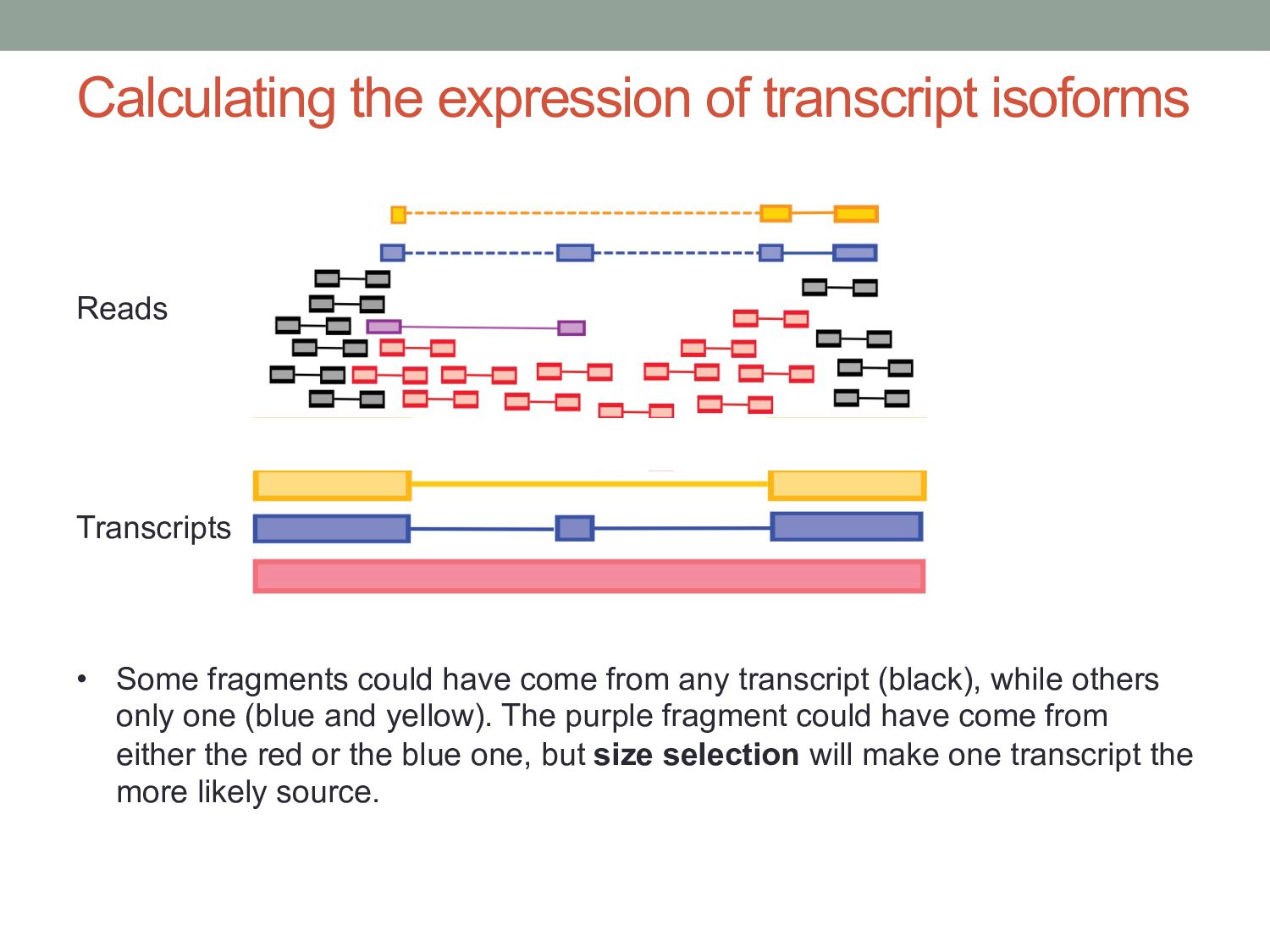
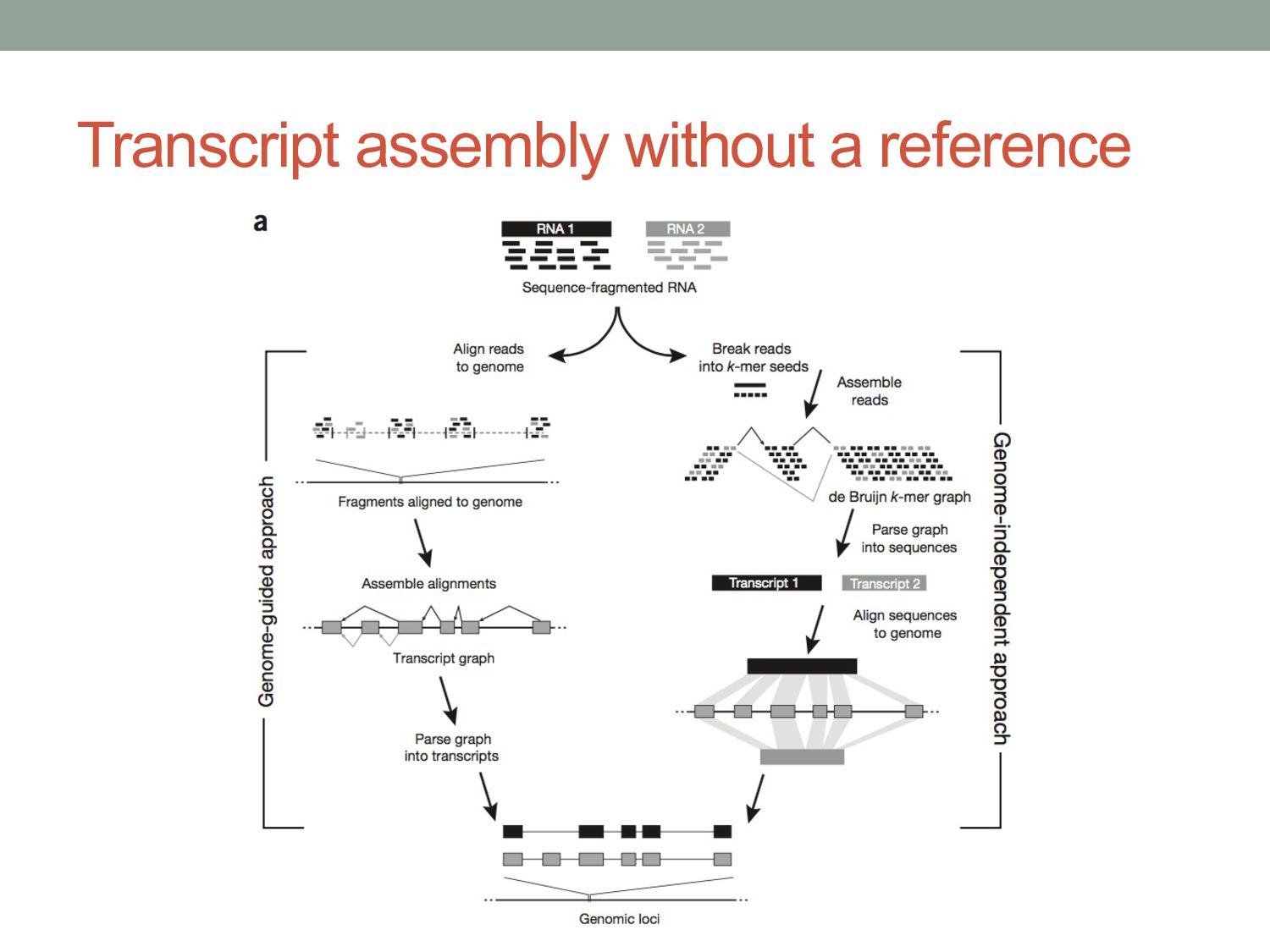
genes represented by only a few reads • Genes can have many isoforms, making it challenging to determine which isoform produced each read. Can RNA-seq tell us which isoforms are expressed?
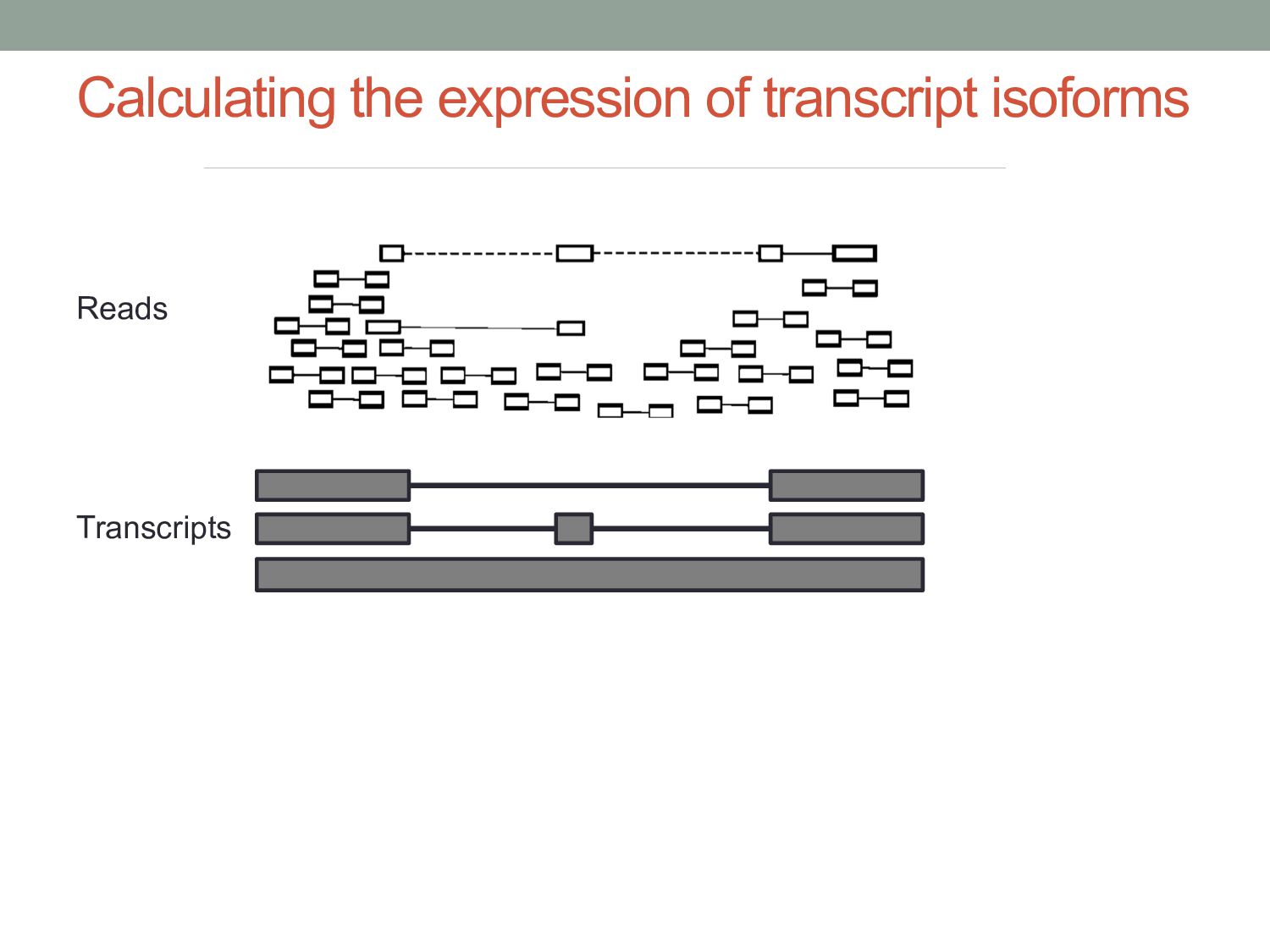
have come from any transcript (black), while others only one (blue and yellow). The purple fragment could have come from either the red or the blue one, but size selection will make one transcript the more likely source. Reads Transcripts
vary across cell types or conditions. With functional genomics assays, this involves analyzing differences in read count information. • Statistical analysis of sequencing (count-based) data needs to account for many sources of variance. • Differentially expressed genes can be found using statistical tests based on the Negative Binomial distribution.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}