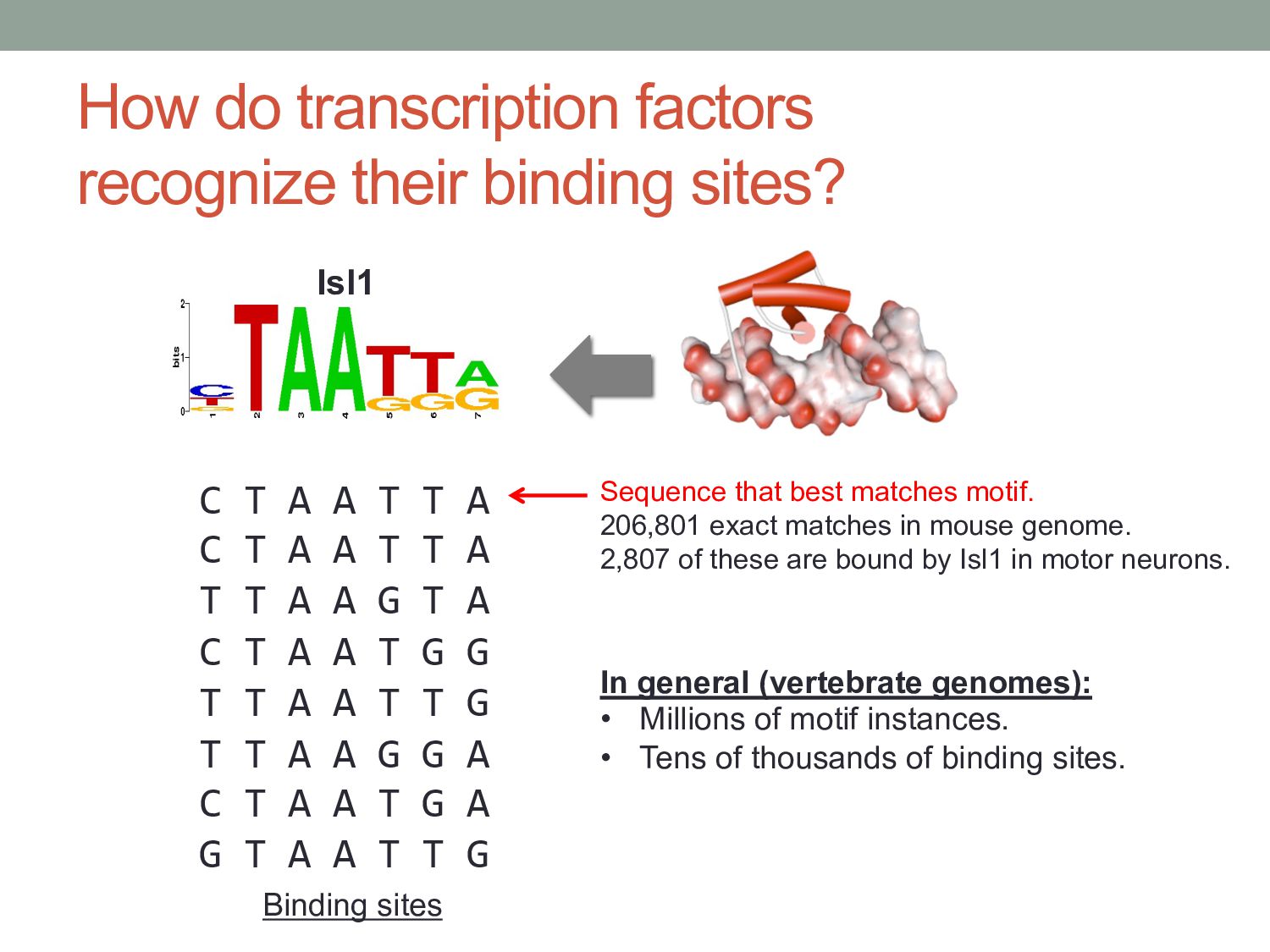
A A T T A C T A A T T A T T A A G T A C T A A T G G T T A A T T G T T A A G G A C T A A T G A G T A A T T G Binding sites Sequence that best matches motif. 206,801 exact matches in mouse genome. 2,807 of these are bound by Isl1 in motor neurons. In general (vertebrate genomes): • Millions of motif instances. • Tens of thousands of binding sites. Isl1
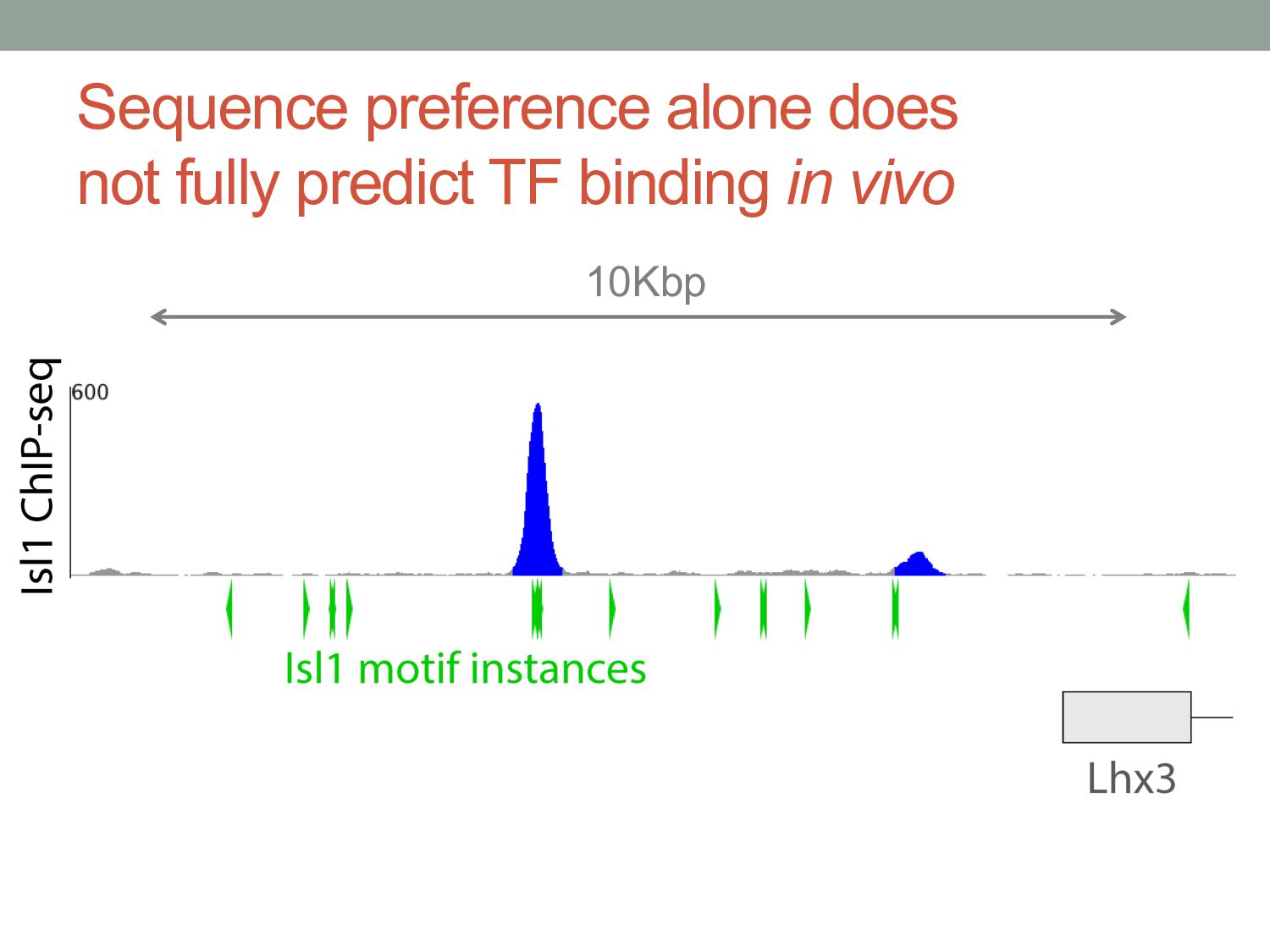
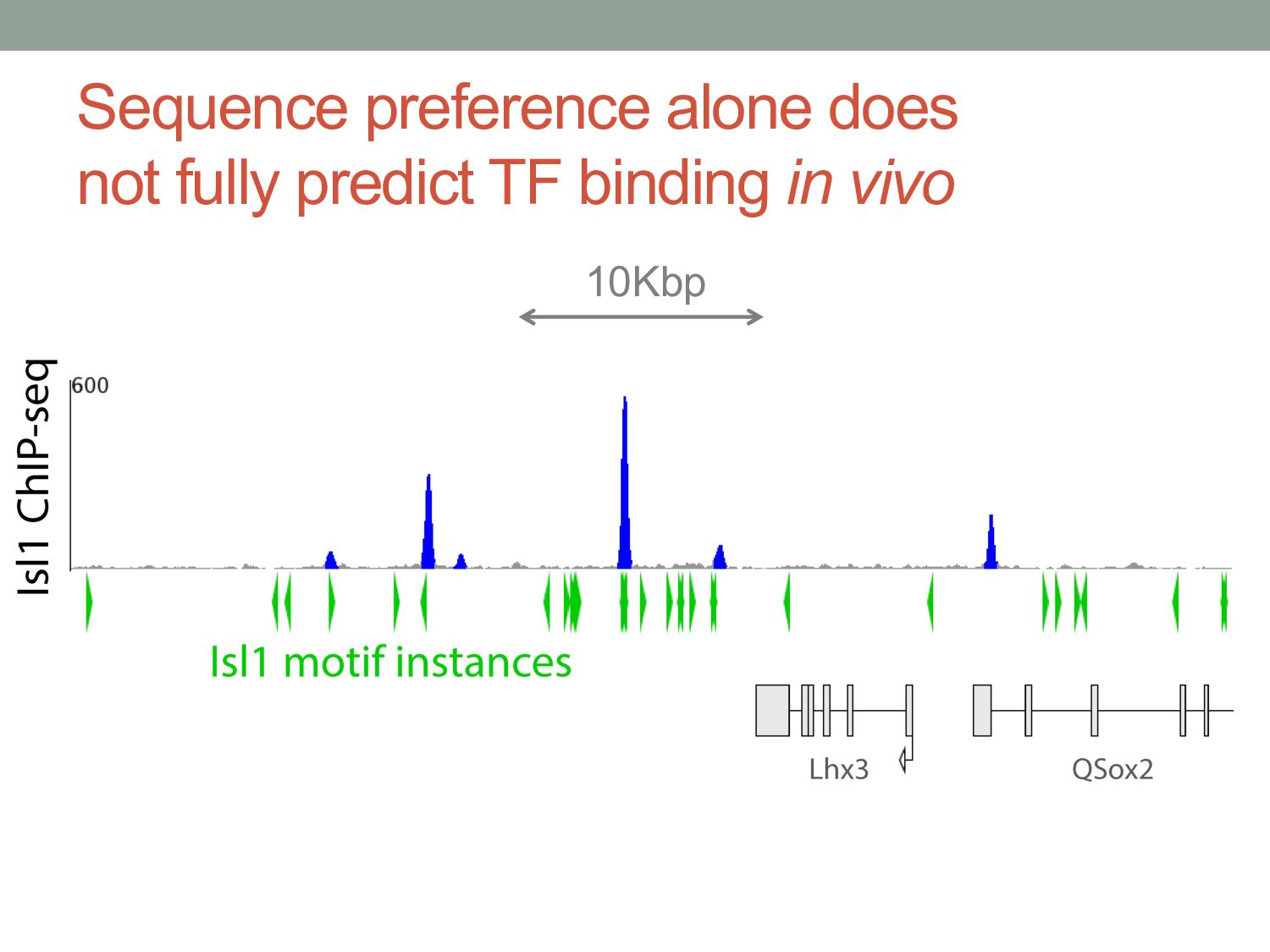
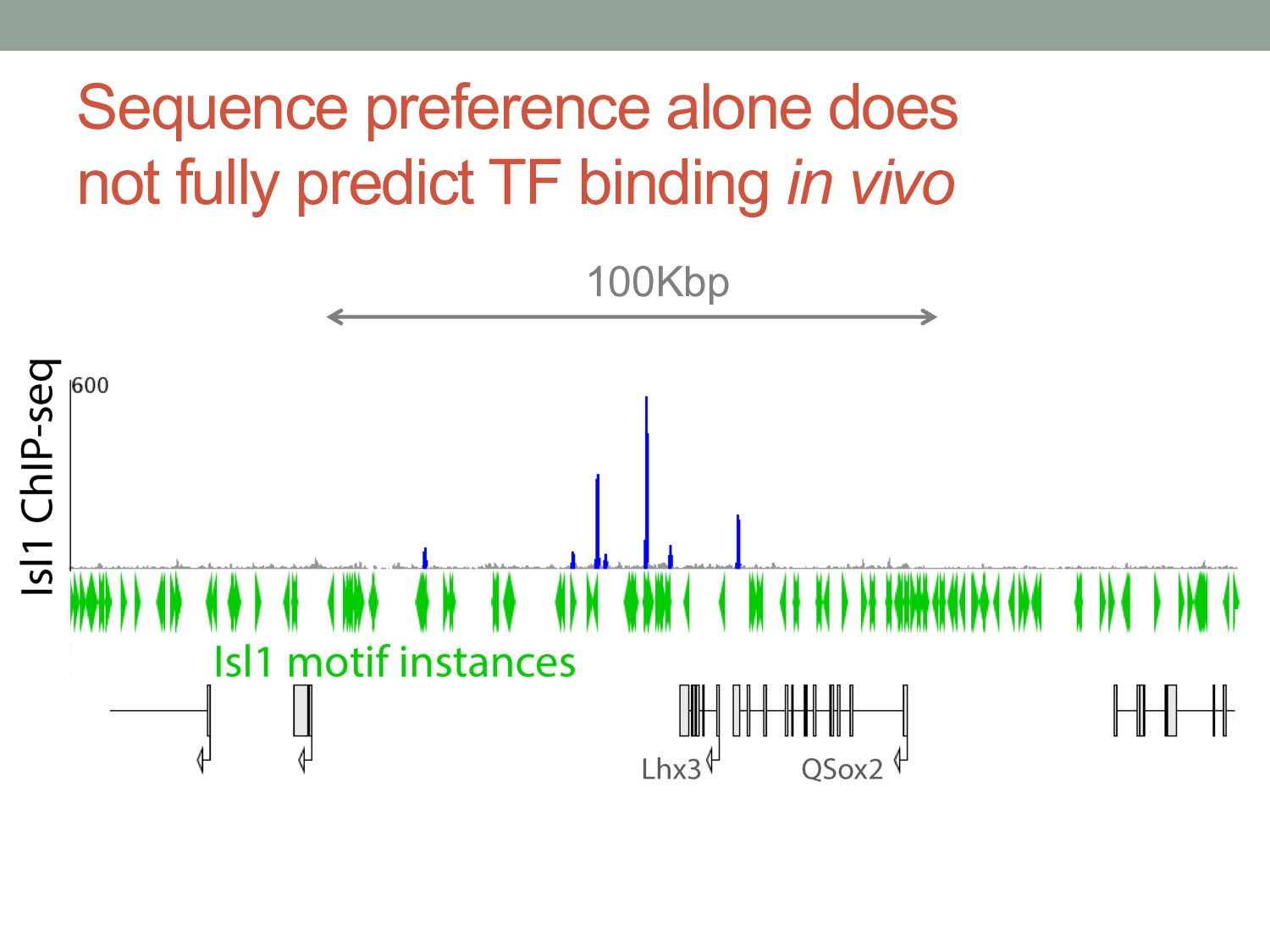
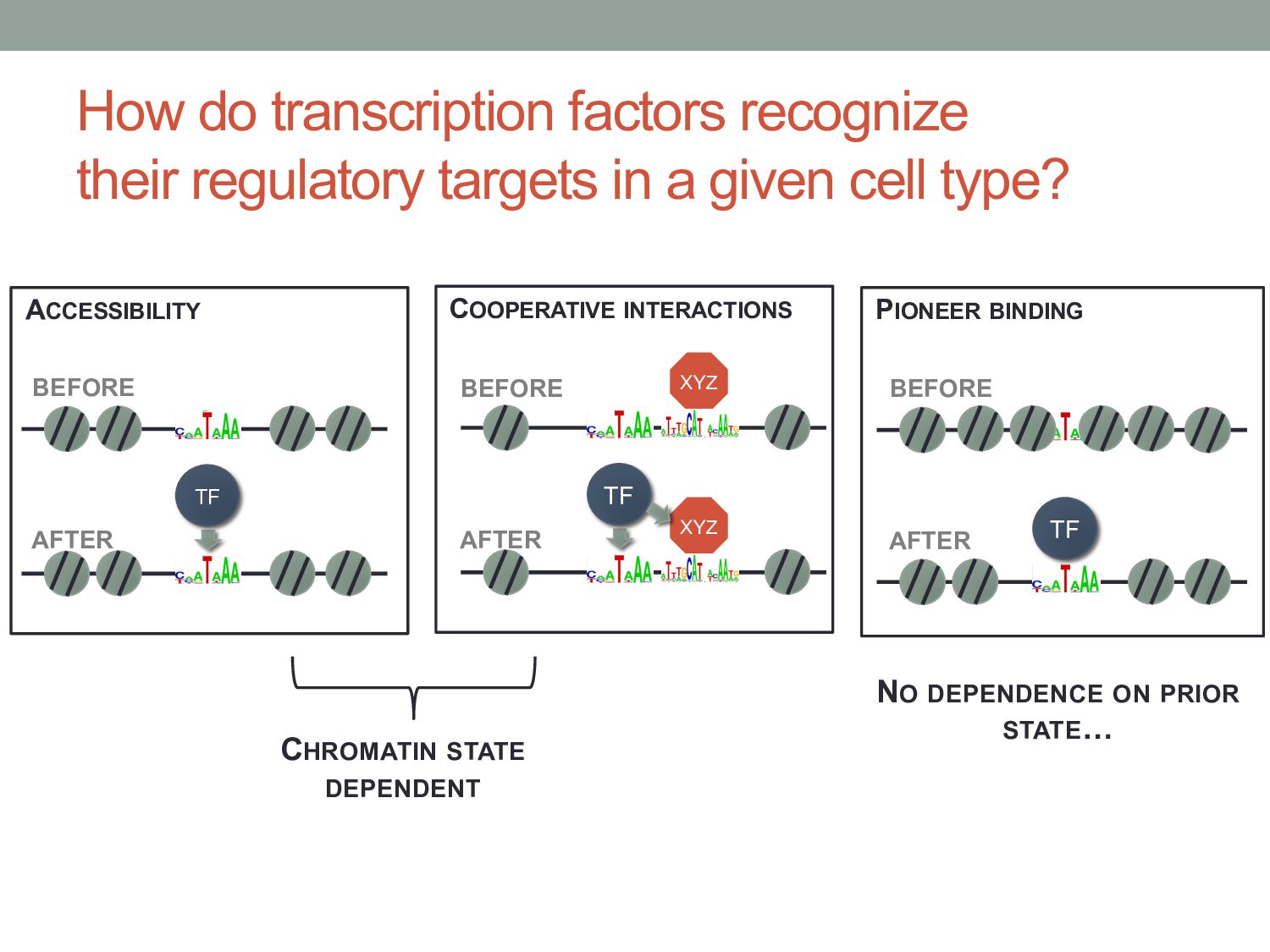
TF binding motif occurrences will have no function in a given cell type*. • How can we focus on motif instances that are more likely to be bound & functional? • Conservation • Cis-regulatory modules (i.e., clusters of sites) • Measuring TF binding (ChIP-seq) • Accessibility (DNaseI hypersensitivity) • Chromatin marks (H3K4me1, H3K27ac) * Wasserman & Sandelin, Nature Reviews Genetics (2004) Cell type dependent: we would need experimental data
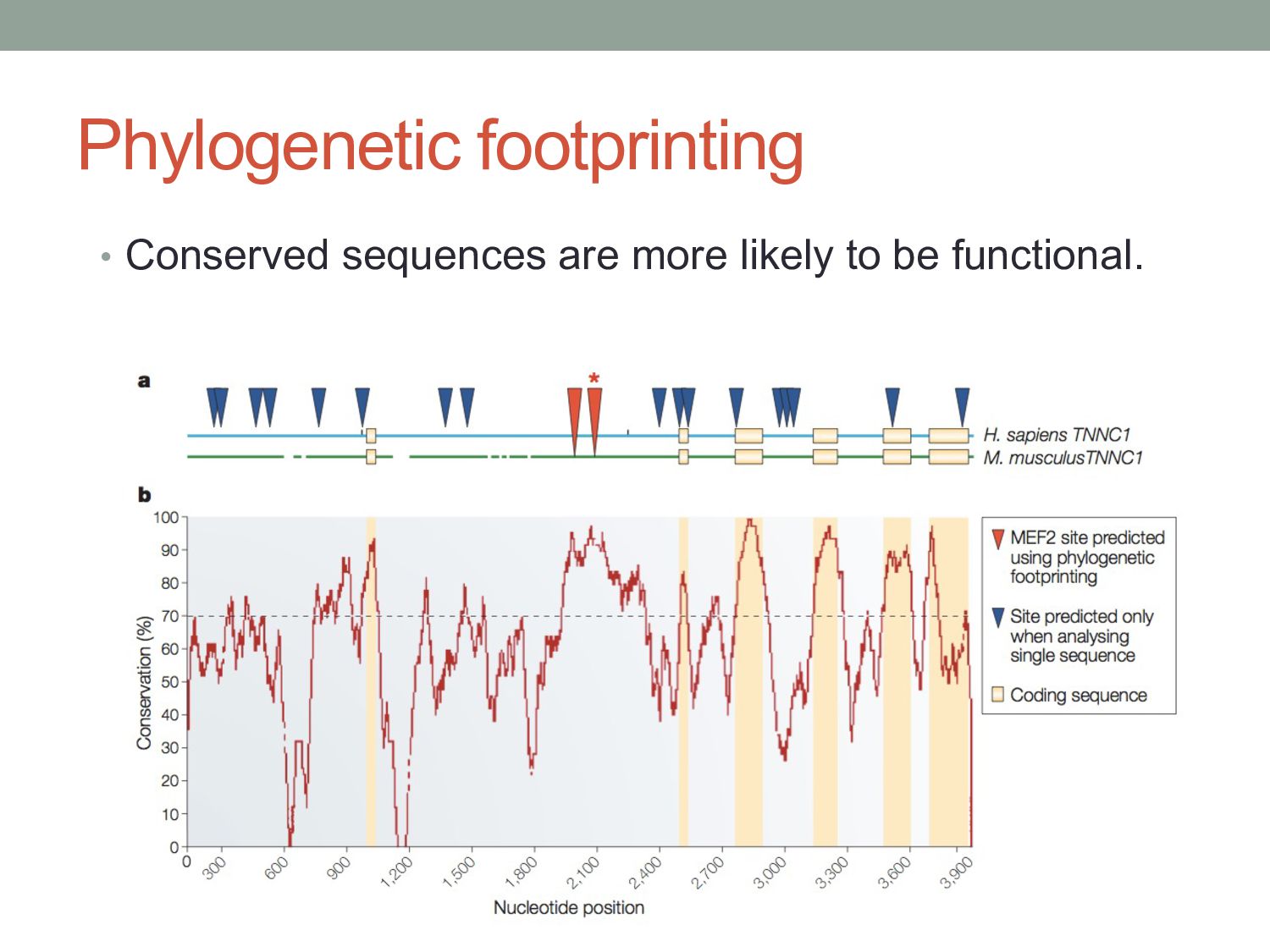
sites are directly conserved between human & mouse. • Perhaps even fewer TF binding sites are functionally conserved. • Phylogenetic shadowing approaches allows conservation analysis of binding sites across more closely related species.
given cell type? CHROMATIN STATE DEPENDENT ACCESSIBILITY TF XYZ COOPERATIVE INTERACTIONS TF XYZ PIONEER BINDING TF BEFORE AFTER BEFORE AFTER BEFORE AFTER NO DEPENDENCE ON PRIOR STATE…
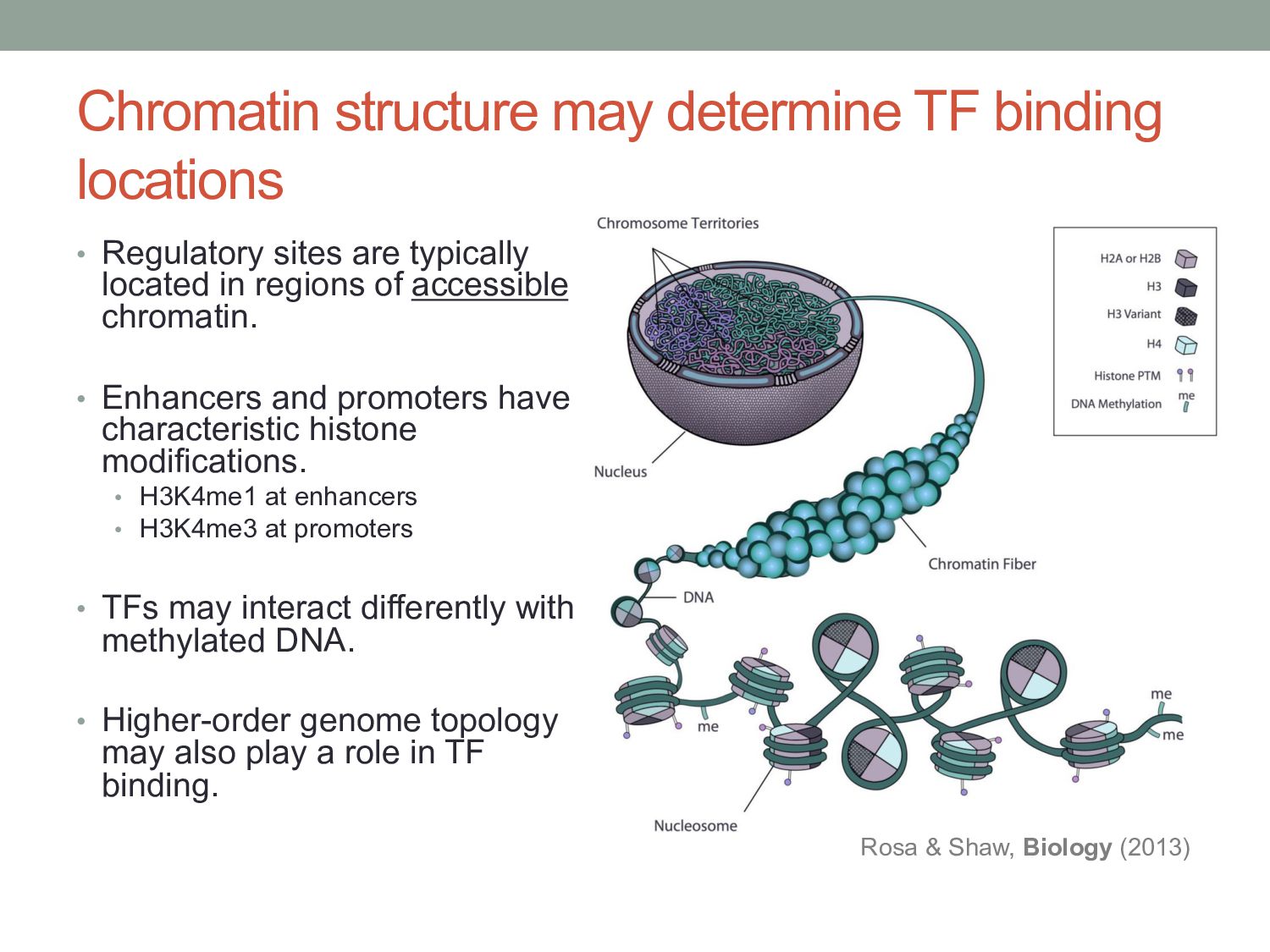
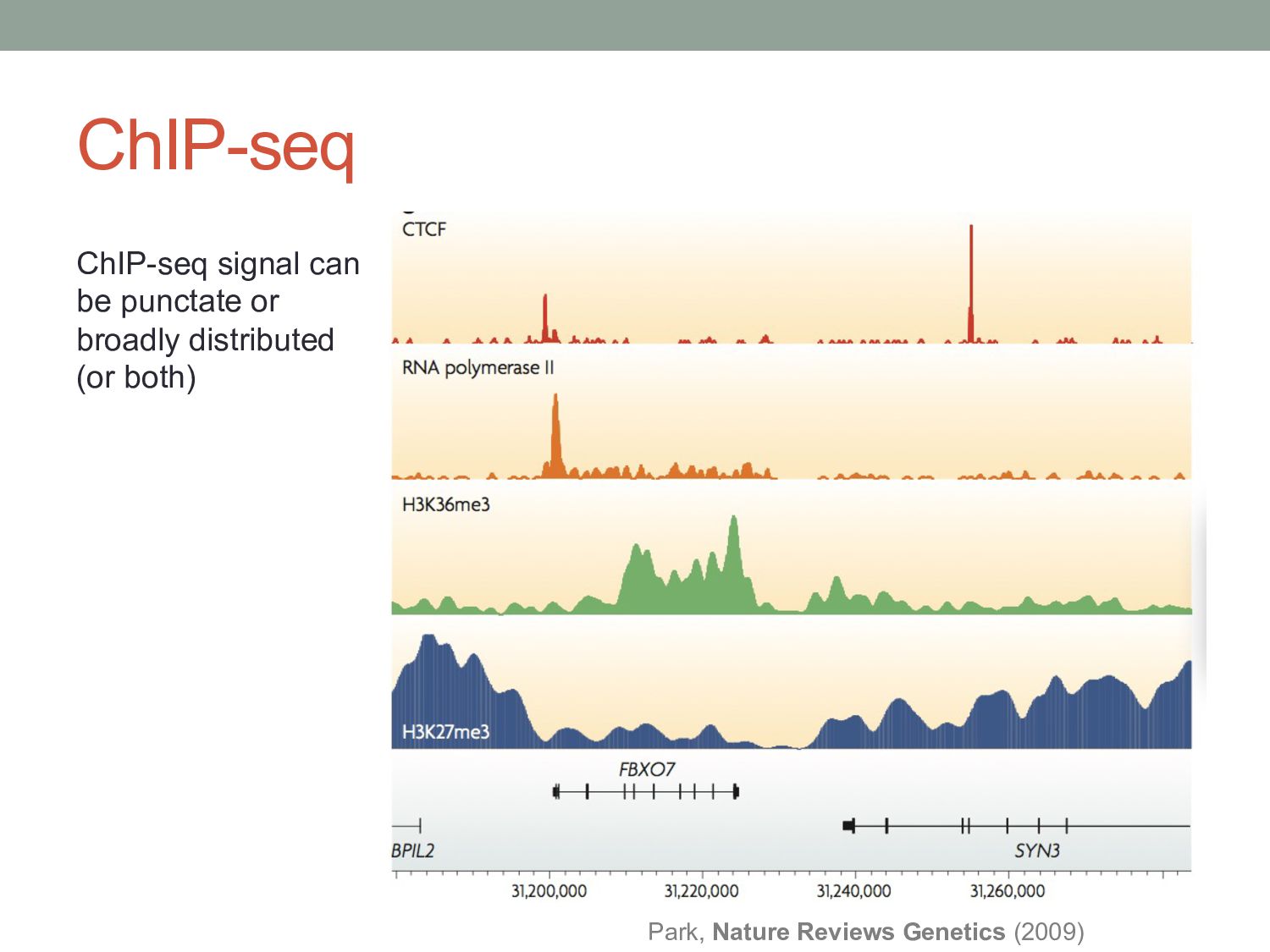
are typically located in regions of accessible chromatin. • Enhancers and promoters have characteristic histone modifications. • H3K4me1 at enhancers • H3K4me3 at promoters • TFs may interact differently with methylated DNA. • Higher-order genome topology may also play a role in TF binding. Rosa & Shaw, Biology (2013)
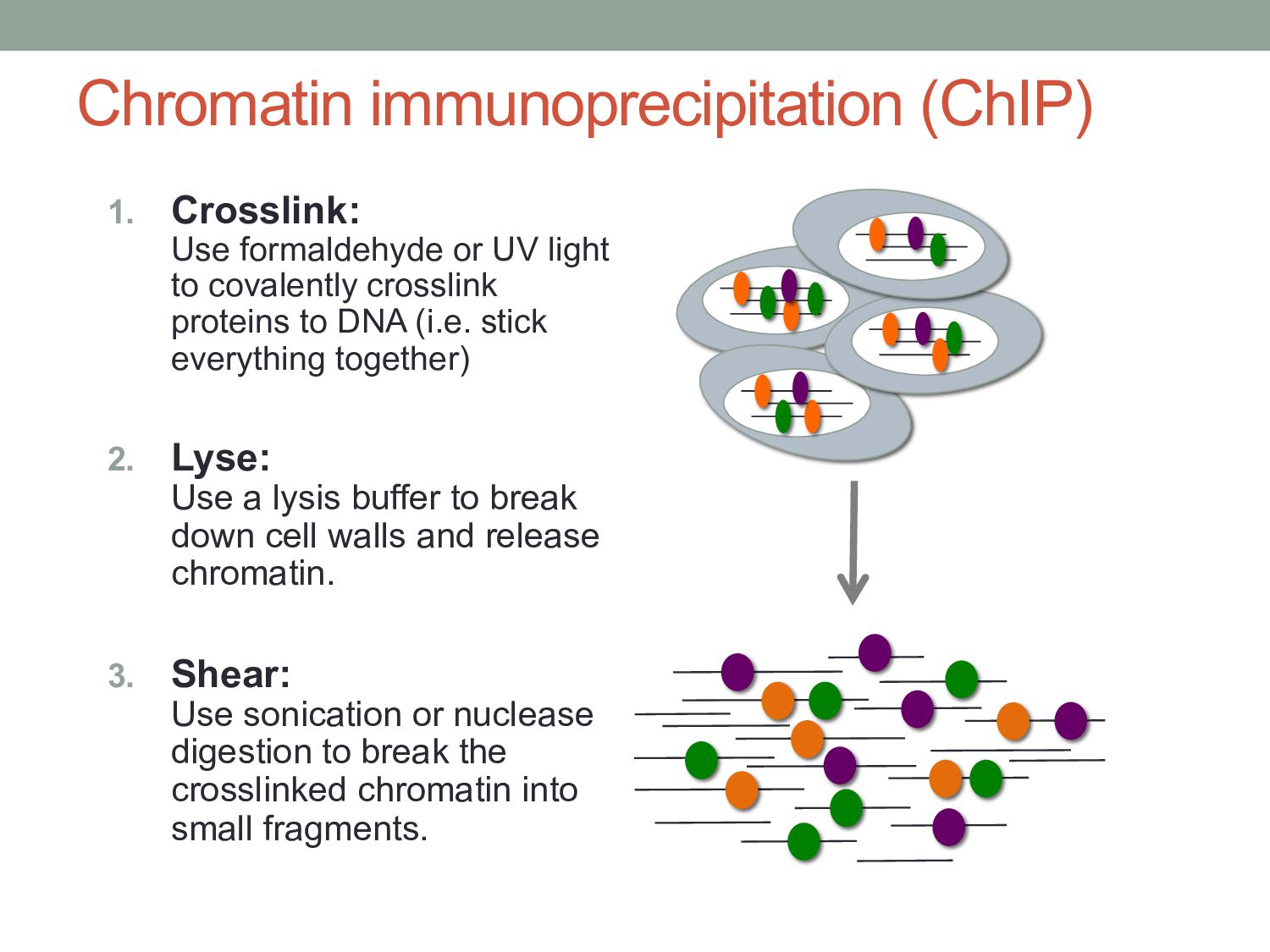
to covalently crosslink proteins to DNA (i.e. stick everything together) 2. Lyse: Use a lysis buffer to break down cell walls and release chromatin. 3. Shear: Use sonication or nuclease digestion to break the crosslinked chromatin into small fragments.
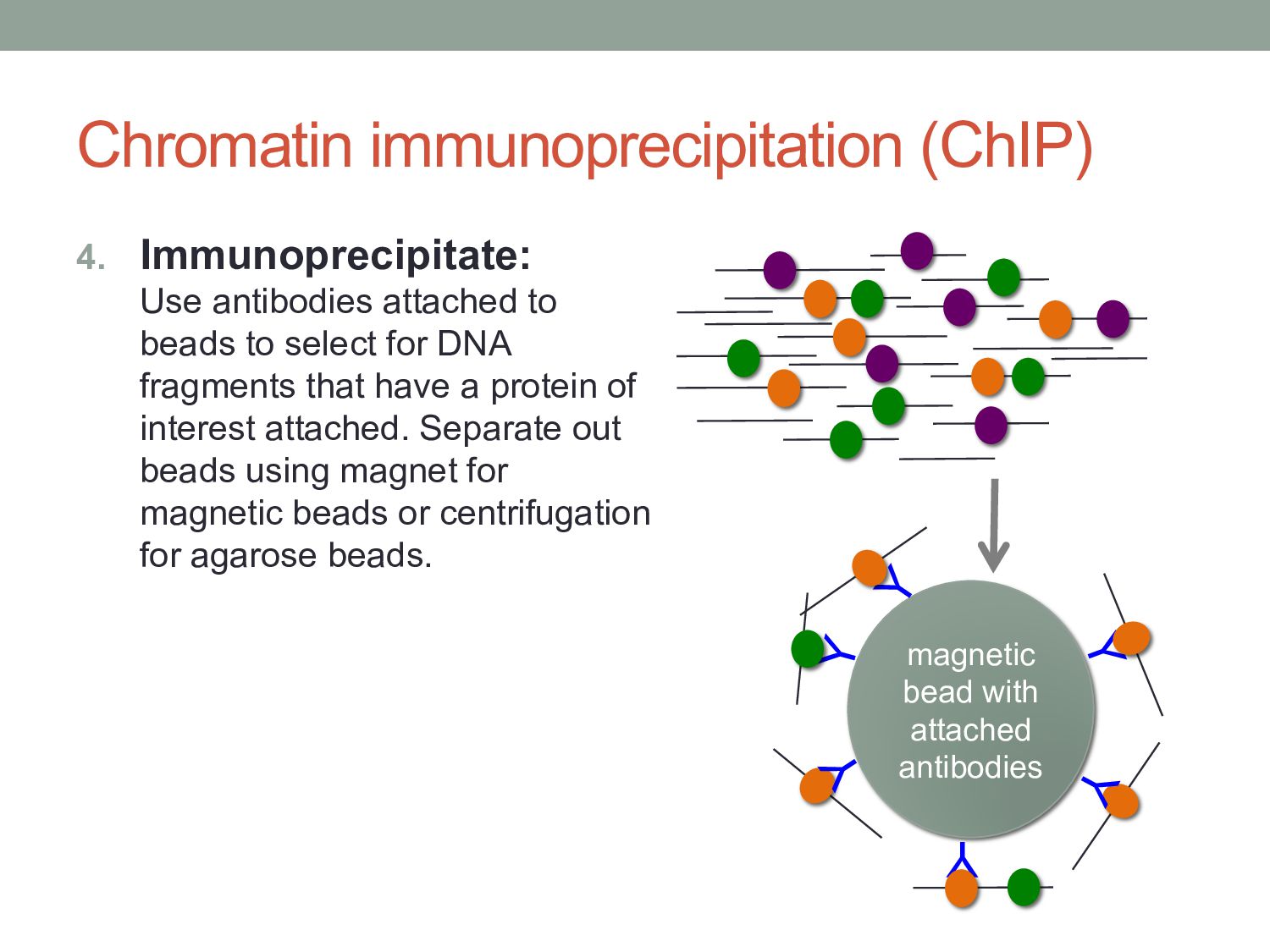
to select for DNA fragments that have a protein of interest attached. Separate out beads using magnet for magnetic beads or centrifugation for agarose beads. Y Y Y Y Y magnetic bead with attached antibodies Y
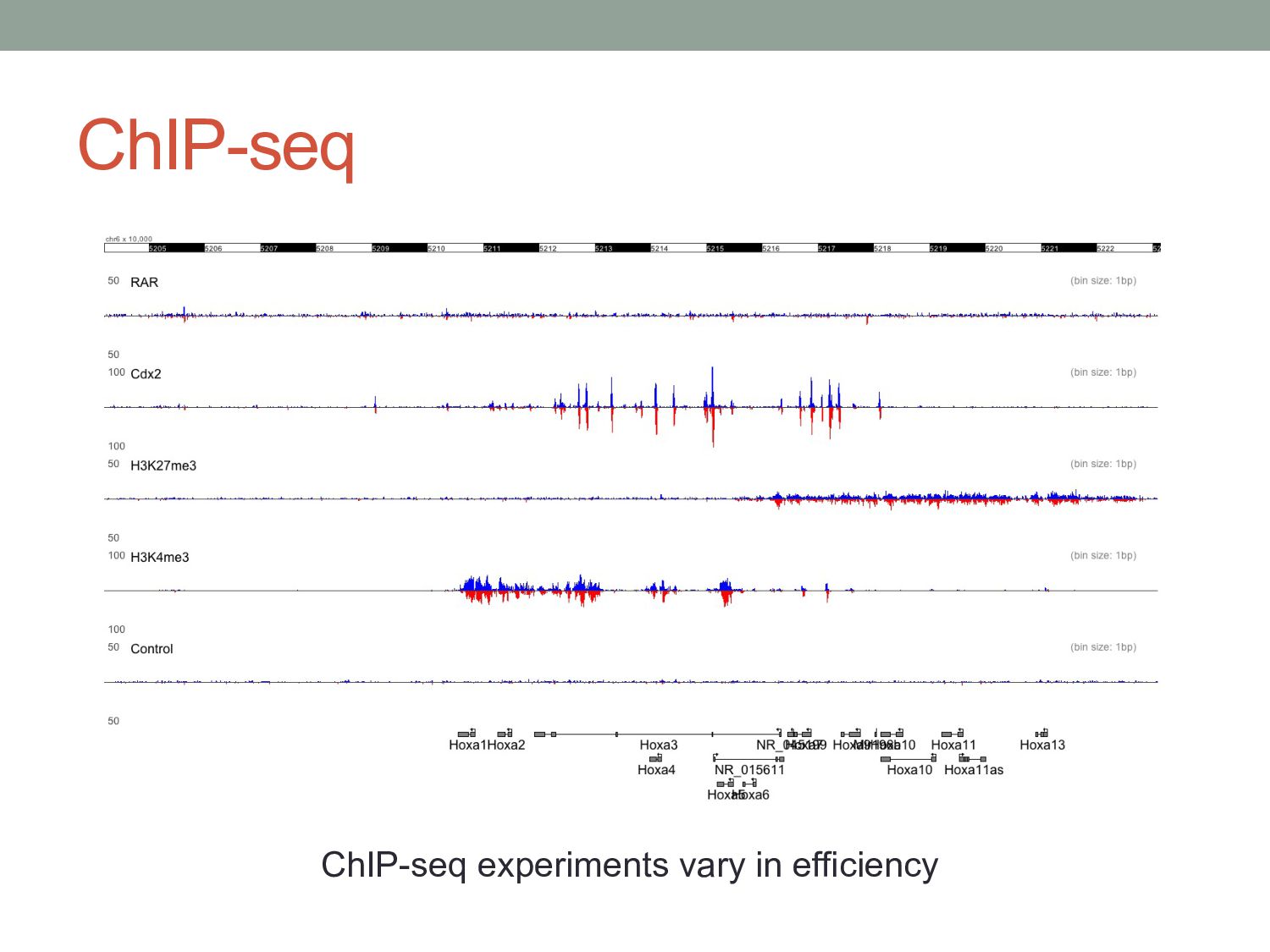
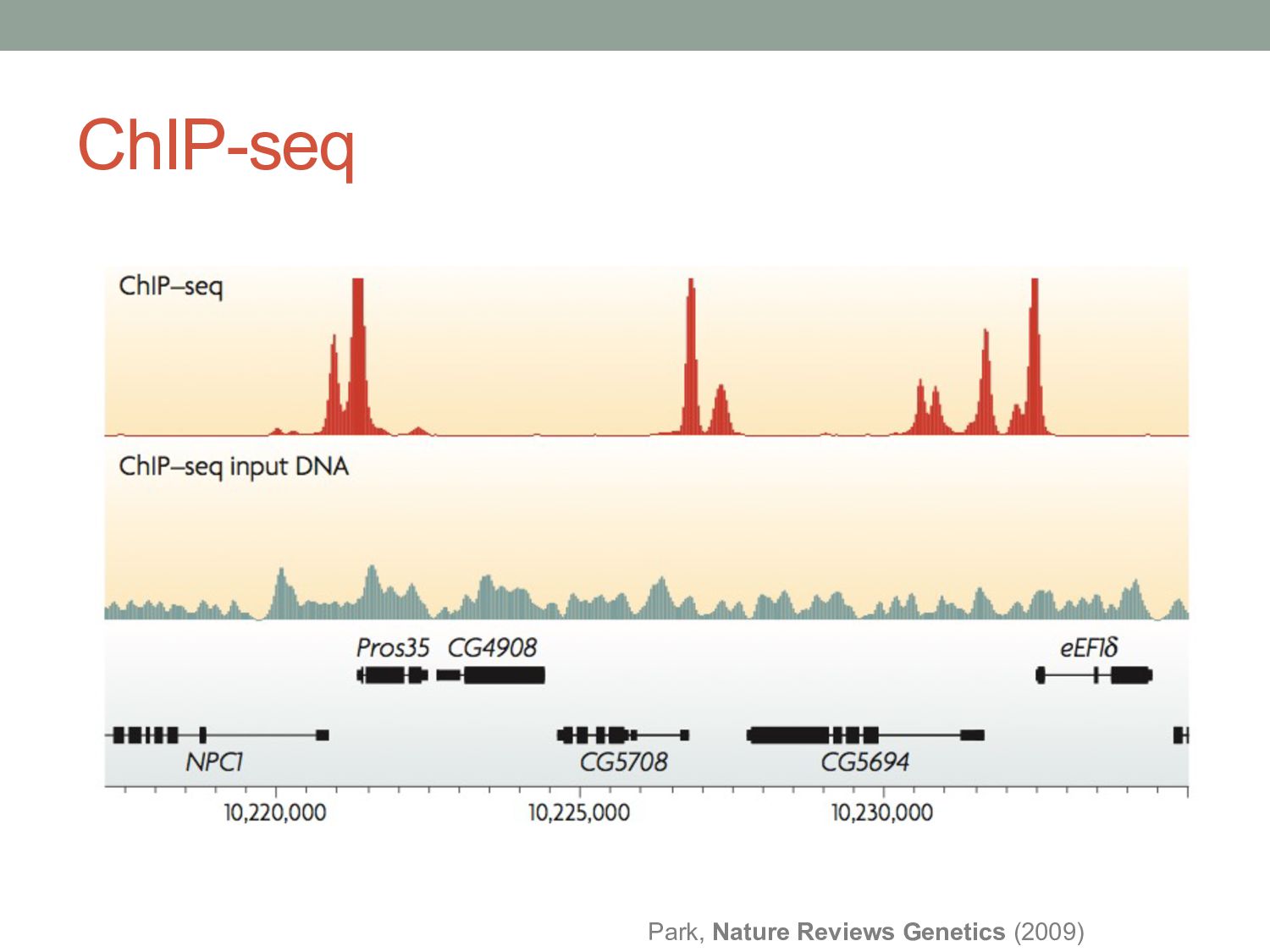
window, looking for regions that have more ChIP-seq reads than we would have expected. • Expected by chance? • Expected according to a control experiment? • What would be a good control experiment? • Sequence input material? • Pseudo IP experiment? • Reads from control experiments are not evenly distributed. • More likely to occur in regions of open chromatin • Possibly more likely to occur near highly expressed genes
expected fragment length, and generate coverage map along genome. • Assume background reads are Poisson distributed. • Mean of the Poisson is locally variable… estimate from control experiment in 5Kbp or 10Kbp around examined location. • For a given location, do we see more reads than we would have expected from the Poisson (p<10-5) MACS: Zhang, et al. Genome Biology, 2008
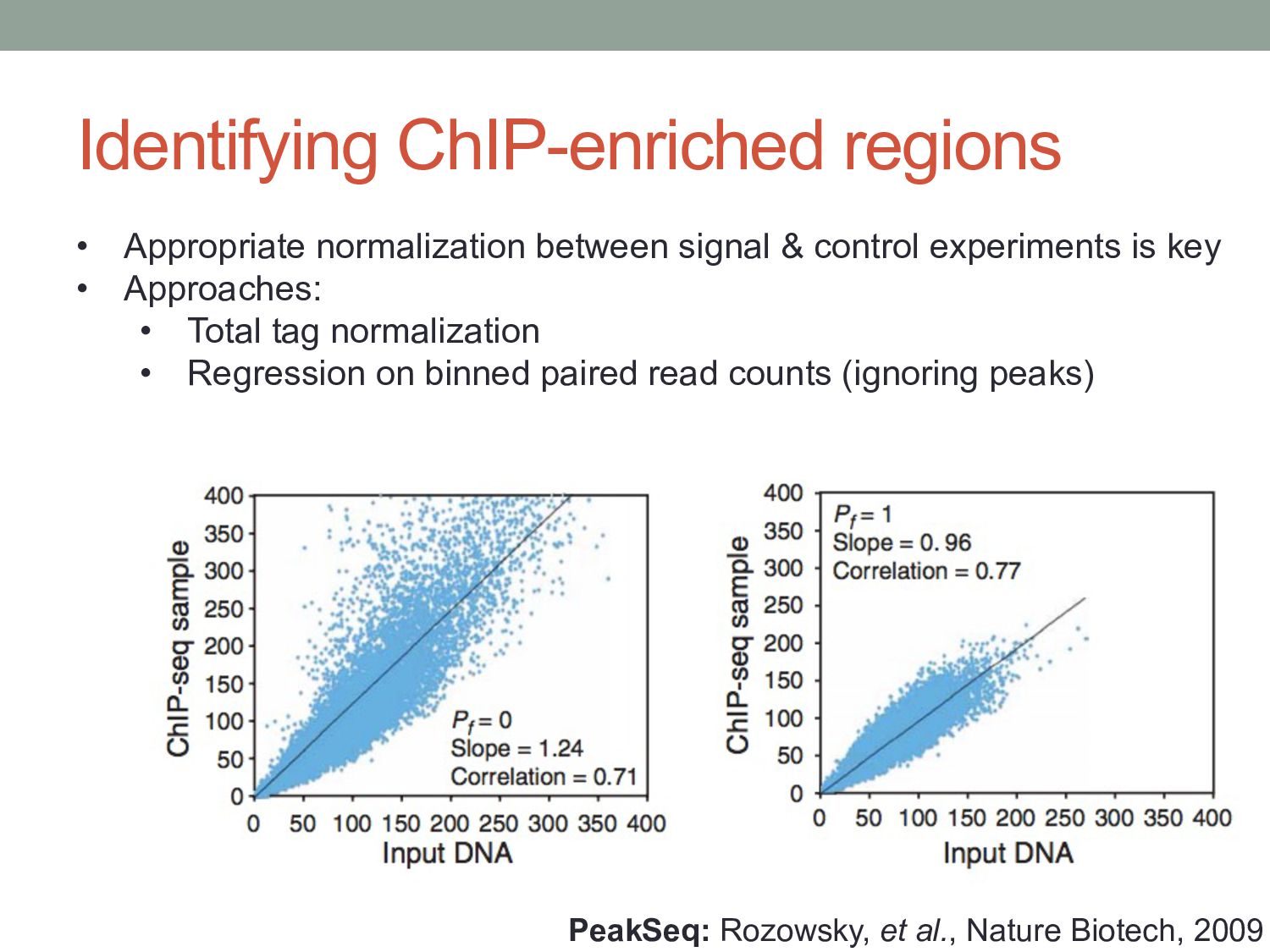
• Appropriate normalization between signal & control experiments is key • Approaches: • Total tag normalization • Regression on binned paired read counts (ignoring peaks)
enriched locations • Examine only the signal experiment. • Determine an enrichment threshold by simulating the same number of reads and setting a false discovery rate. • Step 2: • Find a control vs. signal scaling ratio by performing regression using non-candidate regions. • Use Binomial test of population proportion differences to assess differences between signal read counts and corresponding scaled control read counts. • Benjamini-Hochberg correction for multiple hypothesis testing. PeakSeq: Rozowsky, et al., Nature Biotech, 2009
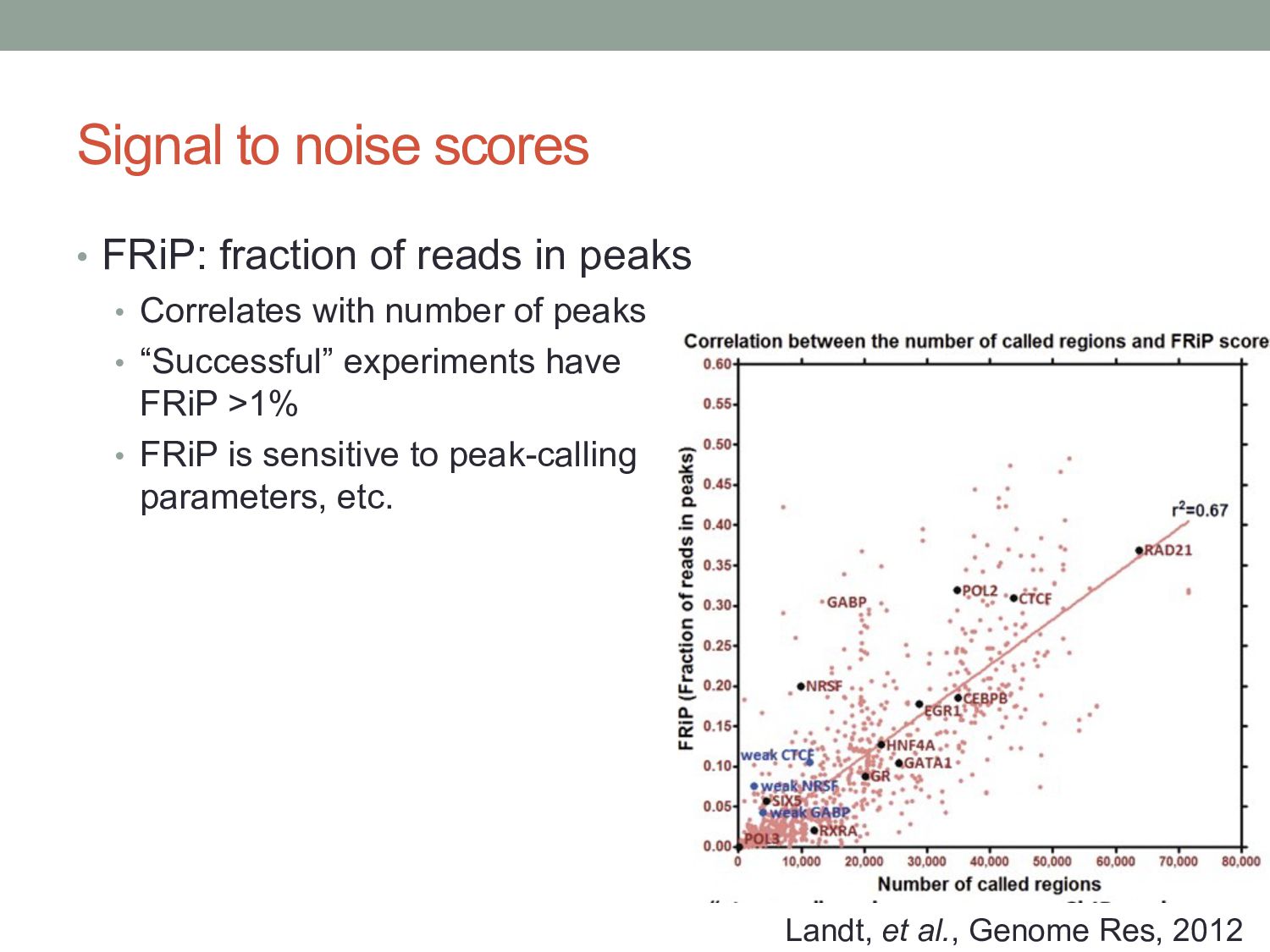
peaks • Correlates with number of peaks • “Successful” experiments have FRiP >1% • FRiP is sensitive to peak-calling parameters, etc. Landt, et al., Genome Res, 2012
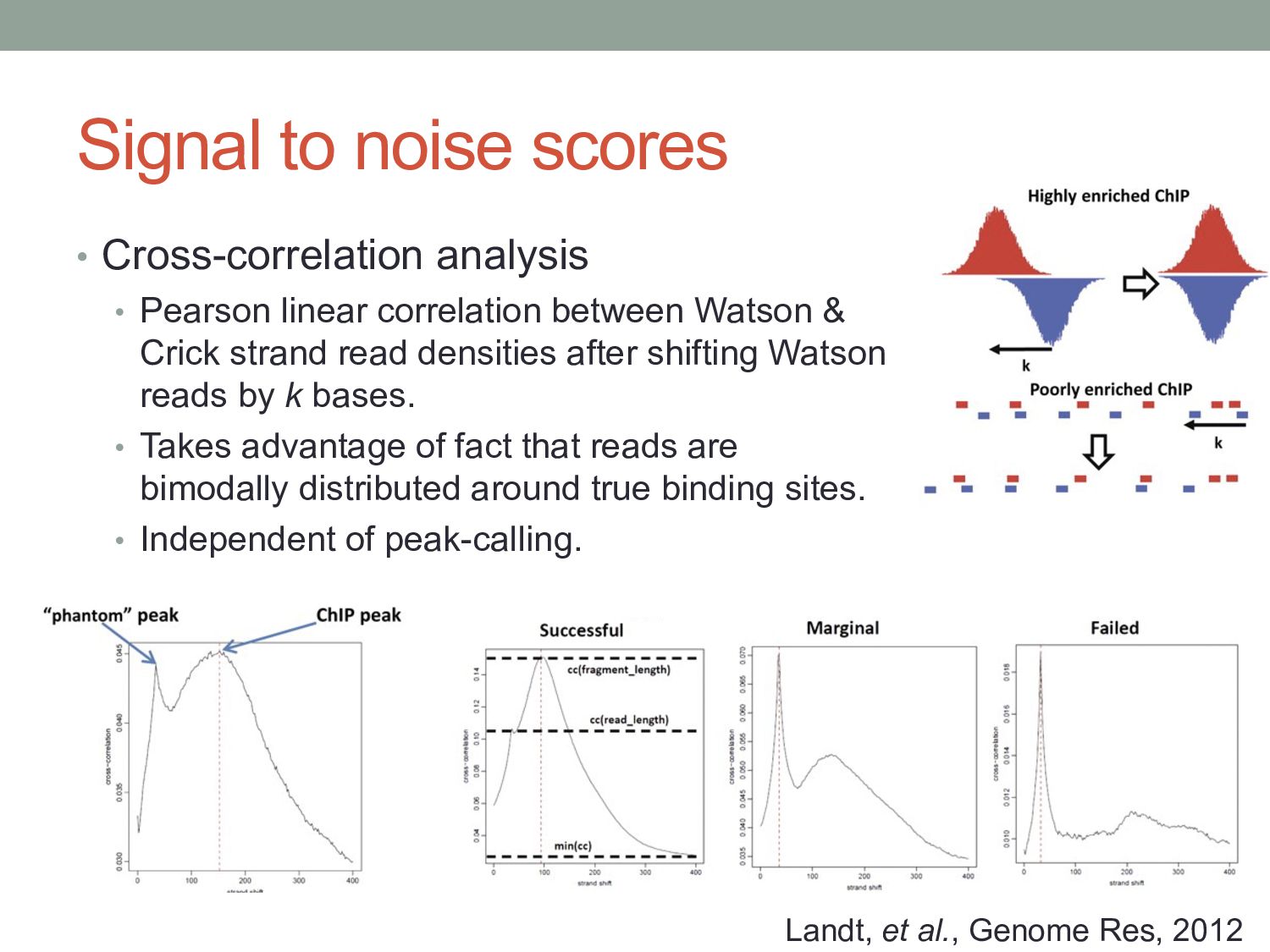
correlation between Watson & Crick strand read densities after shifting Watson reads by k bases. • Takes advantage of fact that reads are bimodally distributed around true binding sites. • Independent of peak-calling. Landt, et al., Genome Res, 2012
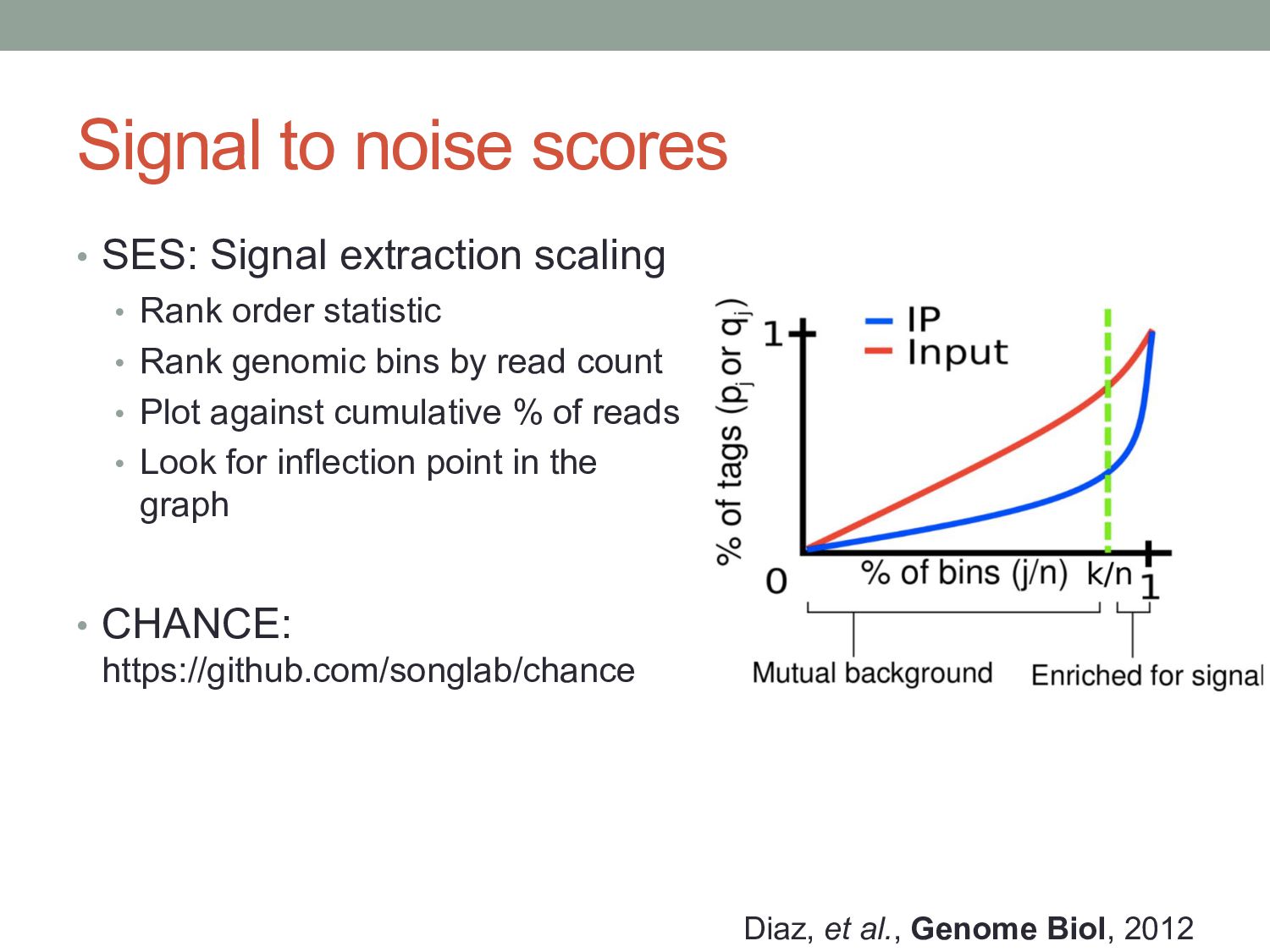
Rank order statistic • Rank genomic bins by read count • Plot against cumulative % of reads • Look for inflection point in the graph • CHANCE: https://github.com/songlab/chance Diaz, et al., Genome Biol, 2012
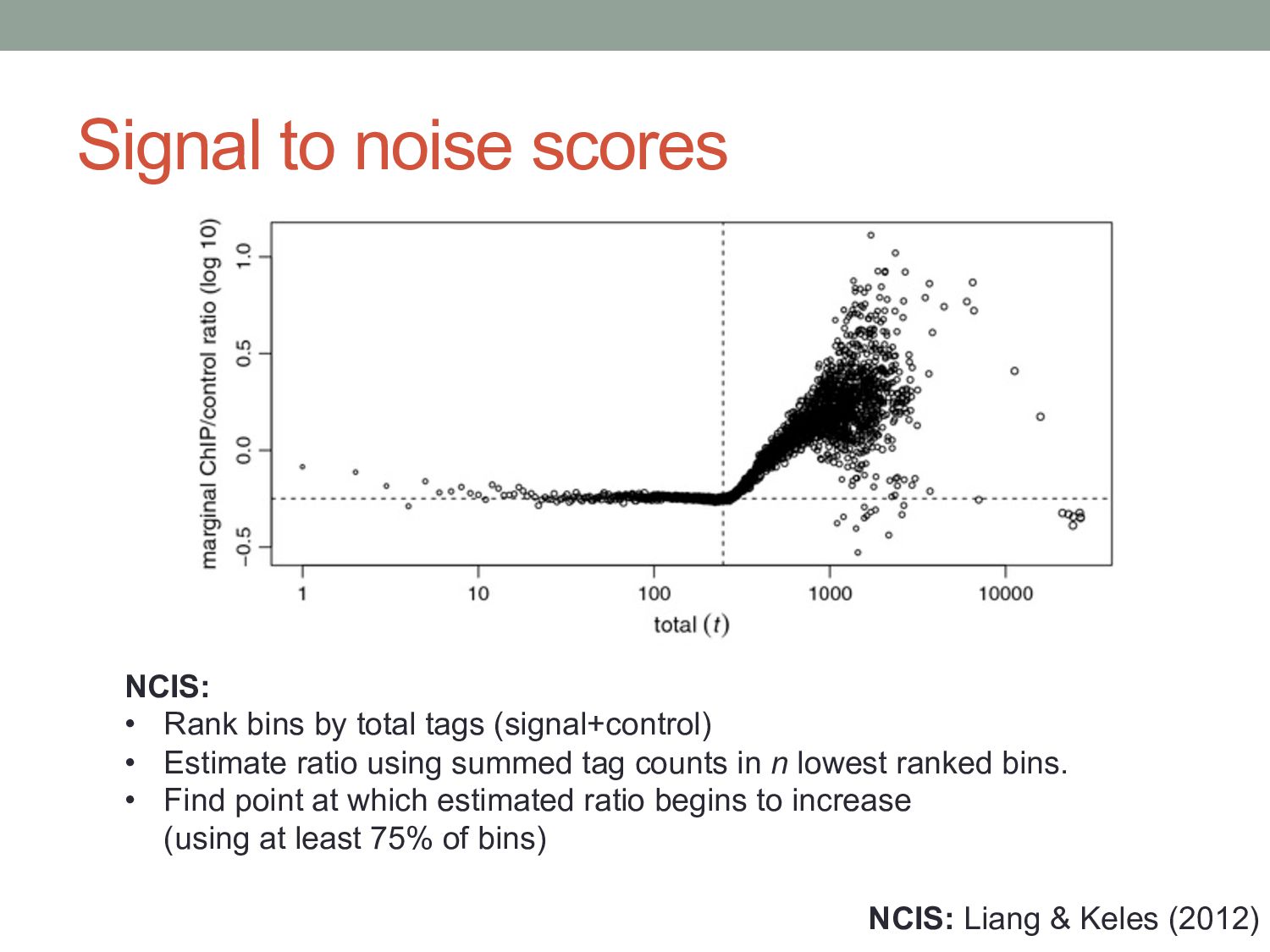
• Rank bins by total tags (signal+control) • Estimate ratio using summed tag counts in n lowest ranked bins. • Find point at which estimated ratio begins to increase (using at least 75% of bins)
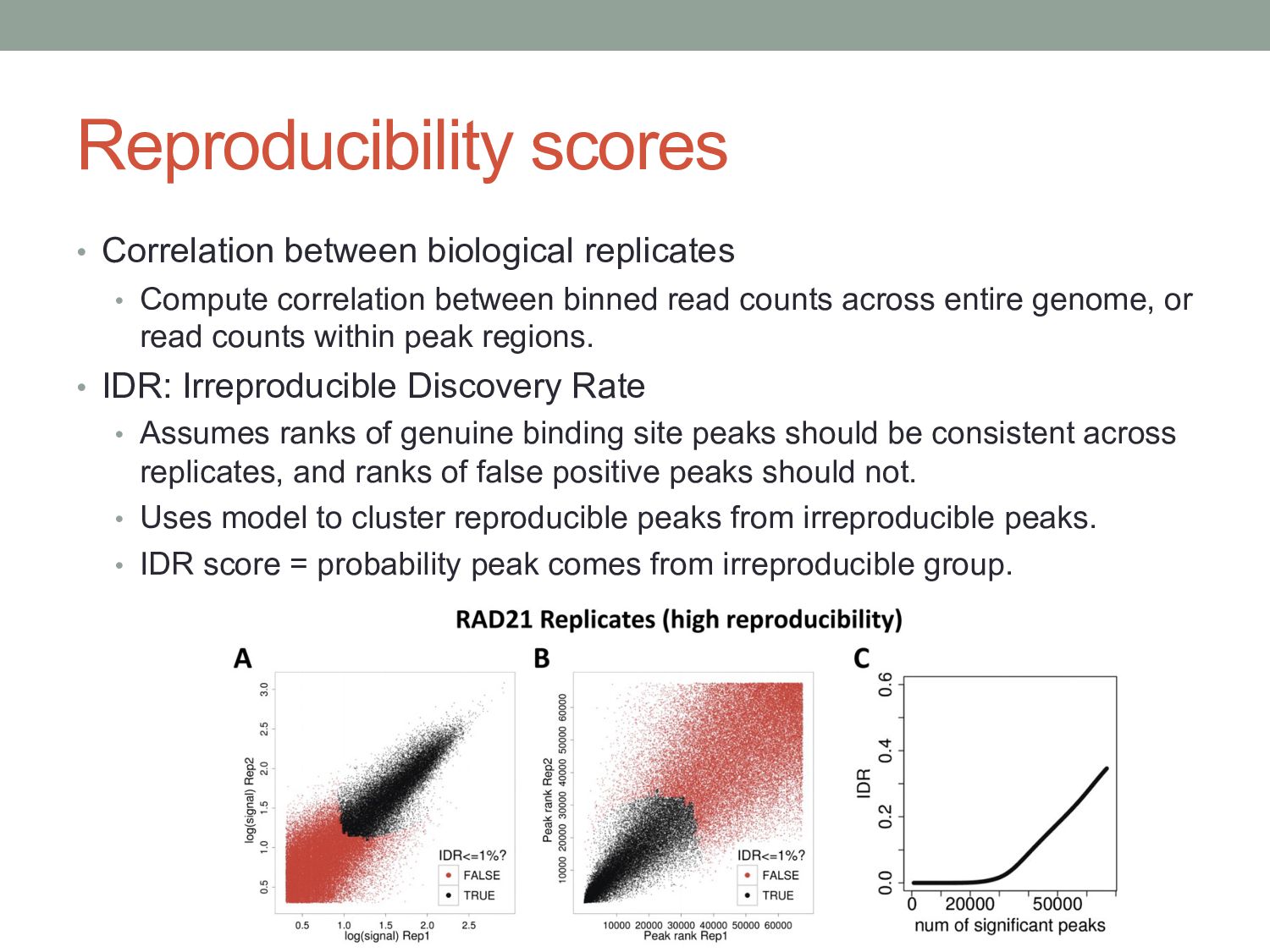
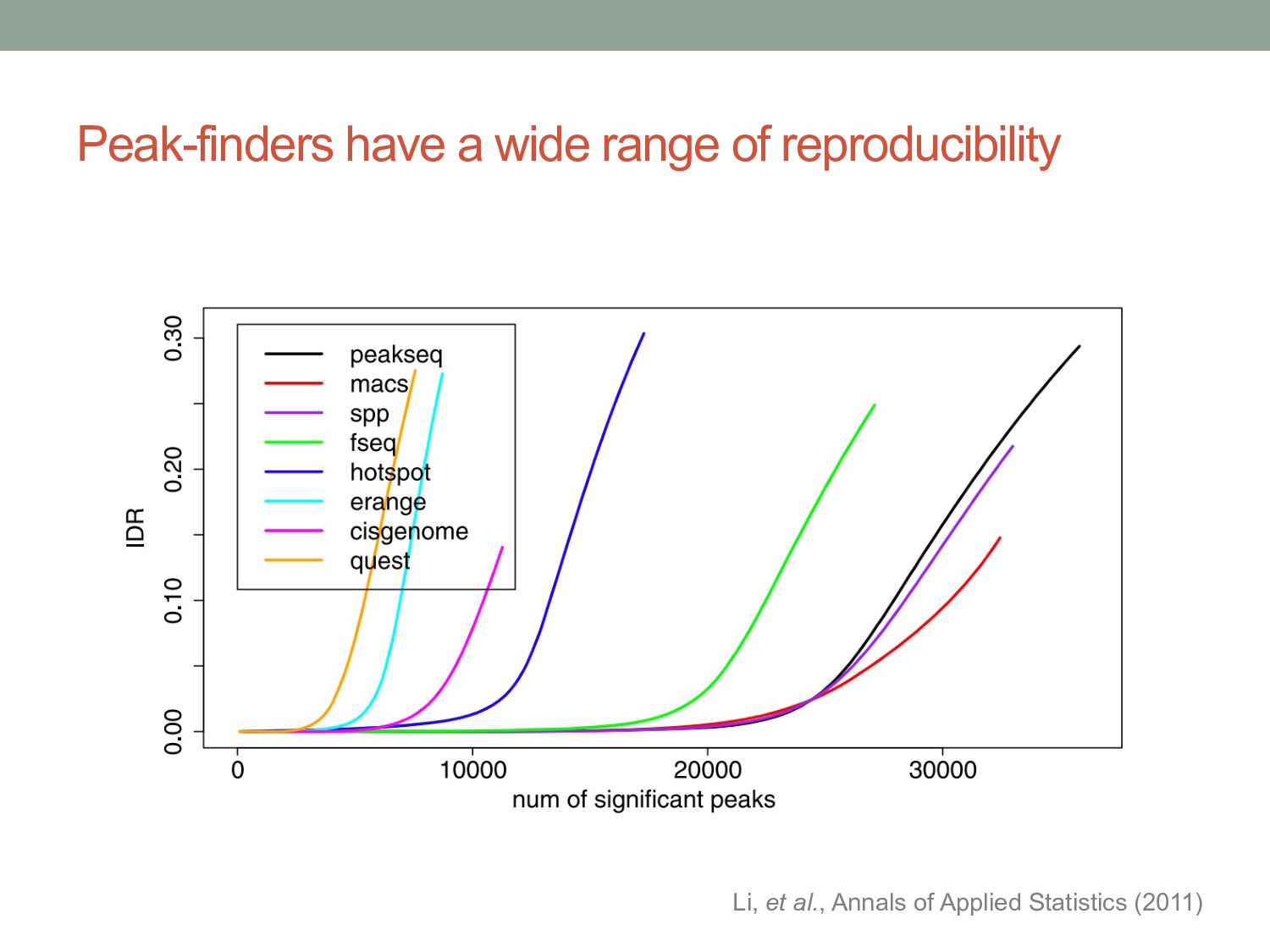
between binned read counts across entire genome, or read counts within peak regions. • IDR: Irreproducible Discovery Rate • Assumes ranks of genuine binding site peaks should be consistent across replicates, and ranks of false positive peaks should not. • Uses model to cluster reproducible peaks from irreproducible peaks. • IDR score = probability peak comes from irreproducible group.
factor will bind in a given cell type. • Motif scanning yields too many potential sites. • ChIP-seq or other experimental approaches are required. • Analysis of ChIP-seq and other protein-DNA binding assay data is not fully standardized • Choice of peak-finding methodology & interpretation of QC metrics is dataset-dependent. • Many tools available for ChIP-seq analysis, only some of which are accessible beyond the command-line. • Few principled performance comparisons have been performed.
a maturing technology”, Nature Reviews Genetics (2009) 10(10):669-680 • Landt S, et al. “ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia”, Genome Research (2012) 22:1813 – 1831 • Carroll TS, et al. “Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data”, Frontiers in Genetics (2014) 5:75 • Mahony S & Pugh BF “Protein-DNA binding in high resolution”, Critical Reviews in Biochemistry and Molecular Biology (2015) 4:269-283
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}