gas Next step: Let’s add a gradient of the density! Generalized gradient approximation (GGA) NANO266 2 E xc GGA[ρ↑,ρ↓]= drρ(r)εxc ( ∫ ρ↑,ρ↓, ∇ρ↑ , ∇ρ↓ )
BLYP, 1988: Exchange by Axel Becke based on energy density of atoms, one parameter + Correlation by Lee-Yang-Parr • PW91, 1991: Perdew-Wang 91Parametrization of real-space cut-off procedure • PBE, 1996: Perdew-Burke-Ernzerhof (re- parametrization and simplification of PW91) • RPBE, 1999: revised PBE, improves surface energetics • PBEsol, 2008: Revised PBE for solids NANO266 3
NANO266 5 E xc meta−GGA[ρ↑,ρ↓]= drρ(r)εxc ( ∫ ρ↑,ρ↓, ∇ρ↑ , ∇ρ↓ ,∇2ρ↑,∇2ρ↓) Tao, J.; Perdew, J. P.; Staroverov, V. N.; Scuseria, G. E. Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids., Phys. Rev. Lett., 2003, 91, 146401, doi:10.1103/PhysRevLett.91.146401.
interaction error • Spurious interaction of the electron not completely cancelled with approximate Exc NANO266 7 E ee = 1 2 ρi (r i )ρj (r j ) r ij dr i dr j ∫∫ E x HF = − 1 2 ρi (r i )ρj (r j ) r ij dr i dr j ∫∫ Includes interaction of electron with itself! HF Exchange cancels self- interaction by construction
most popular functional in quantum chemistry (the 8th most cited paper in all fields) • Originally fitted from a set of atomization energies, ionization potentials, proton affinities and total atomic energies. PBE0: HSE (Heyd-Scuseria-Ernzerhof) (2006): • Effectively PBE0, but with an adjustable parameter controlling the range of the exchange interaction. Hence, known as a screened hybrid functional • Works remarkably well for extended systems like solids NANO266 8 E xc B3LYP = E x LDA + a o (E x HF − E x LDA )+ a x (E x GGA − E x LDA )+ E c LDA +(E c GGA − E c LDA ) where a 0 = −0.20, a x = 0.72, a c = 0.81 E xc PBE0 = 1 4 E x HF + 3 4 E x PBE + E c PBE E xc HSE = aE x HF,SR (ω)+(1− a)E x PBE,SR (ω)+ E x PBE,LR (ω)+ E c PBE a = 1 4 , ω = 0.2
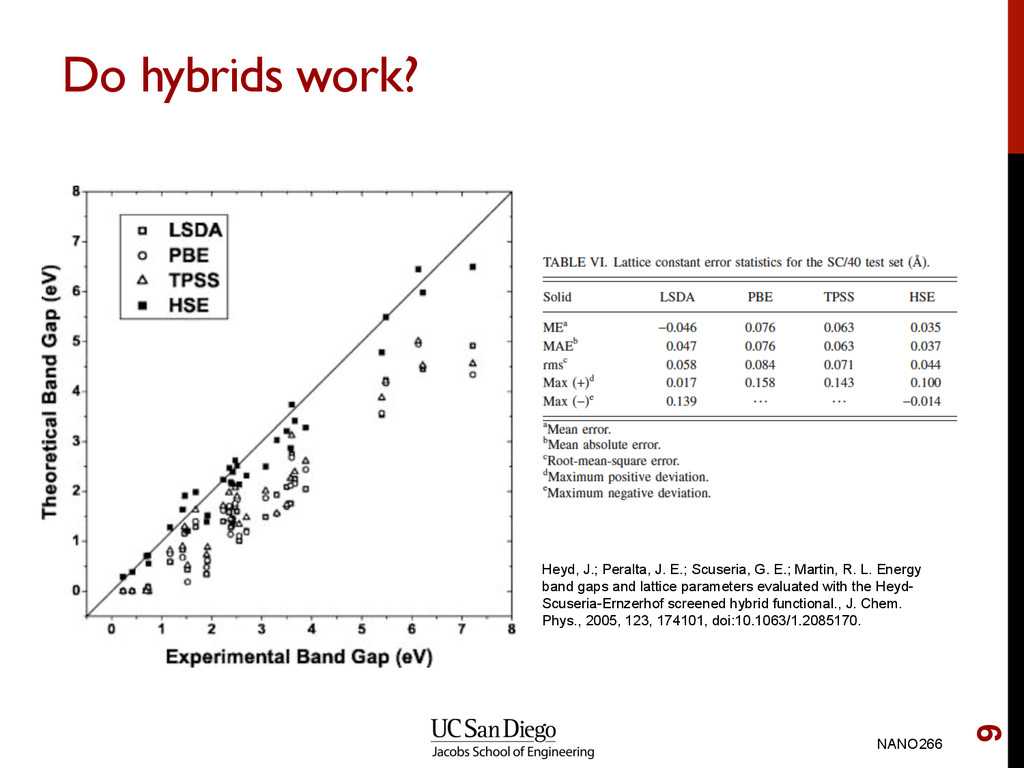
Scuseria, G. E.; Martin, R. L. Energy band gaps and lattice parameters evaluated with the Heyd- Scuseria-Ernzerhof screened hybrid functional., J. Chem. Phys., 2005, 123, 174101, doi:10.1063/1.2085170.
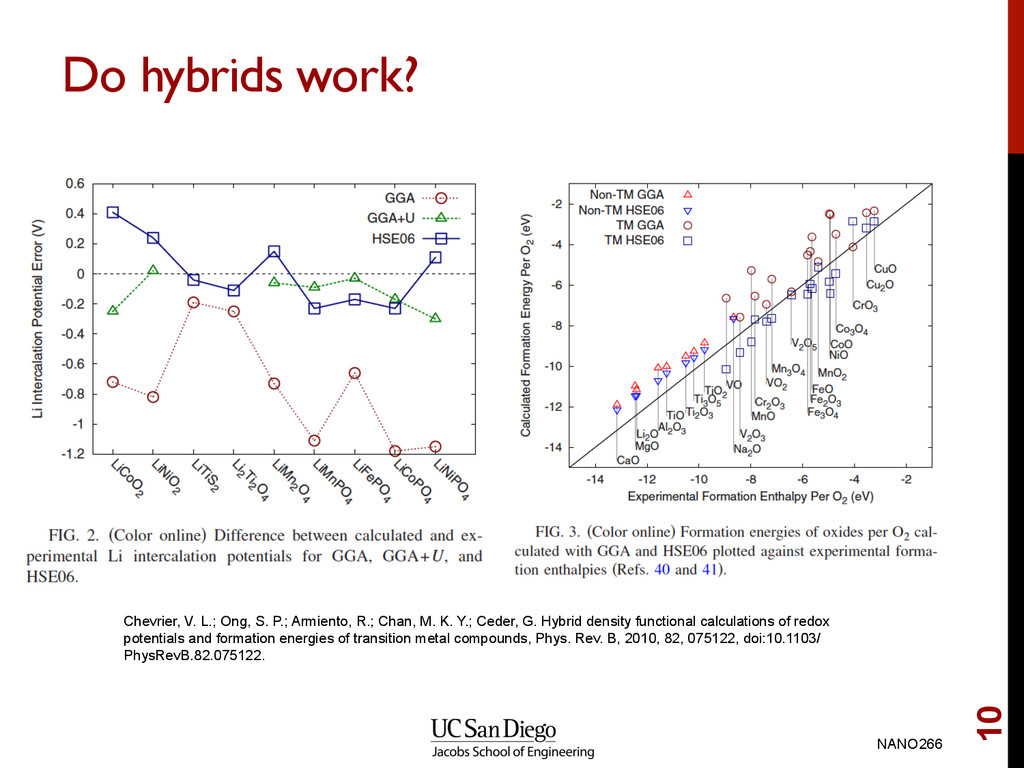
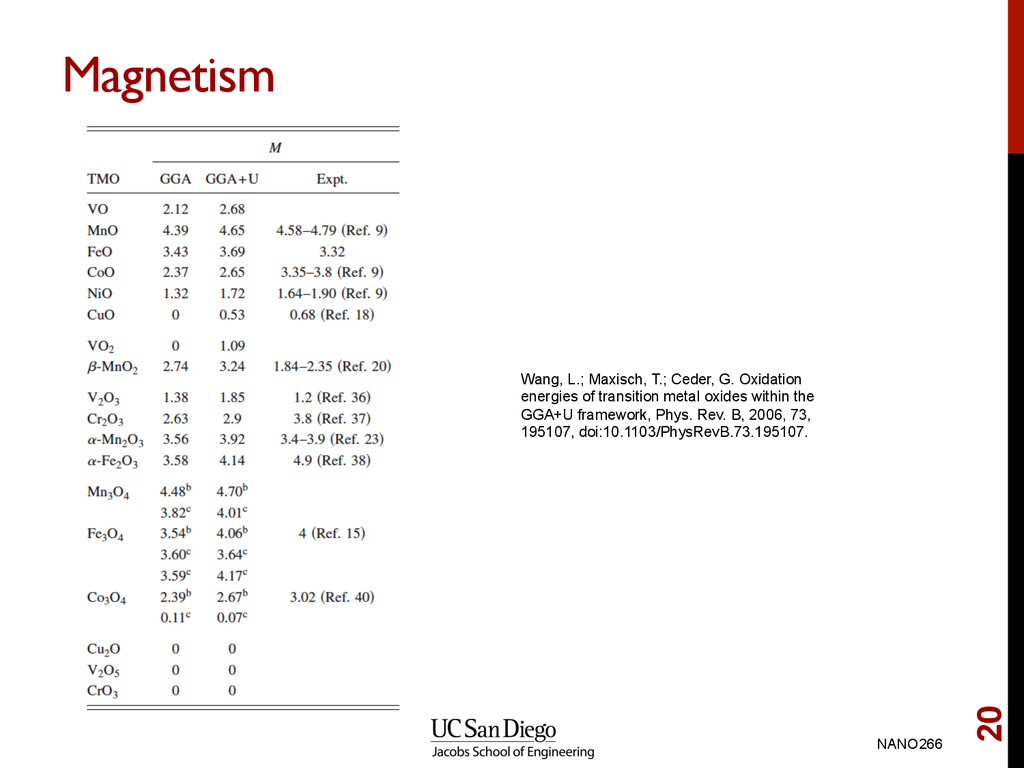
P.; Armiento, R.; Chan, M. K. Y.; Ceder, G. Hybrid density functional calculations of redox potentials and formation energies of transition metal compounds, Phys. Rev. B, 2010, 82, 075122, doi:10.1103/ PhysRevB.82.075122.
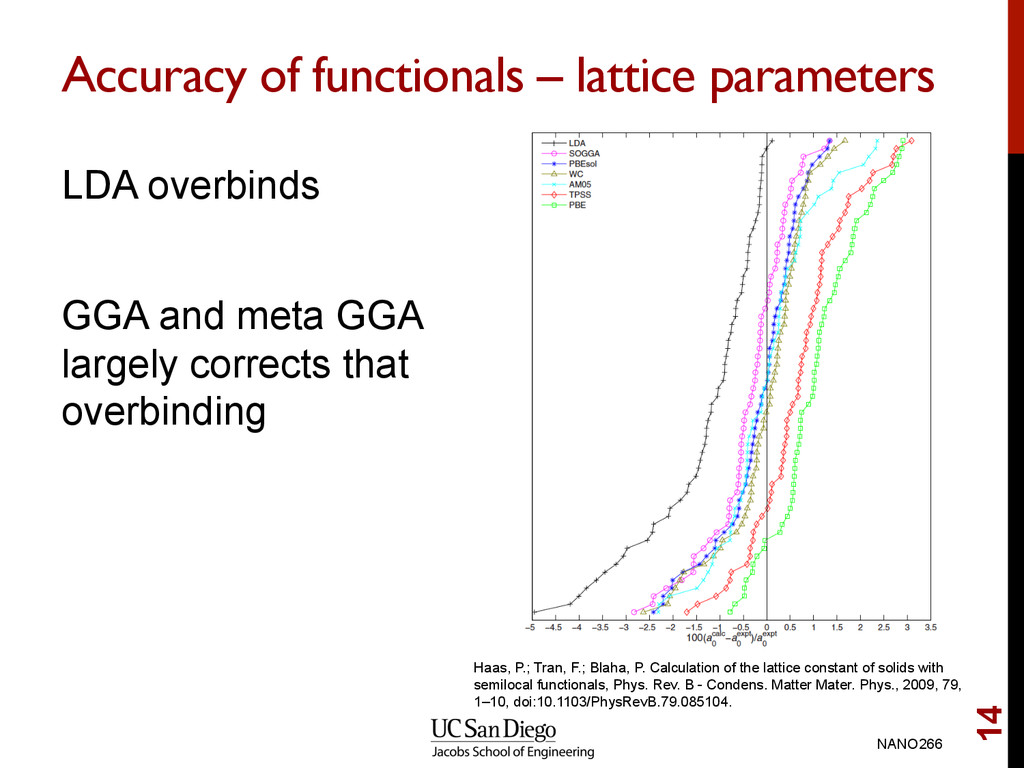
meta GGA largely corrects that overbinding NANO266 14 Haas, P.; Tran, F.; Blaha, P. Calculation of the lattice constant of solids with semilocal functionals, Phys. Rev. B - Condens. Matter Mater. Phys., 2009, 79, 1–10, doi:10.1103/PhysRevB.79.085104.
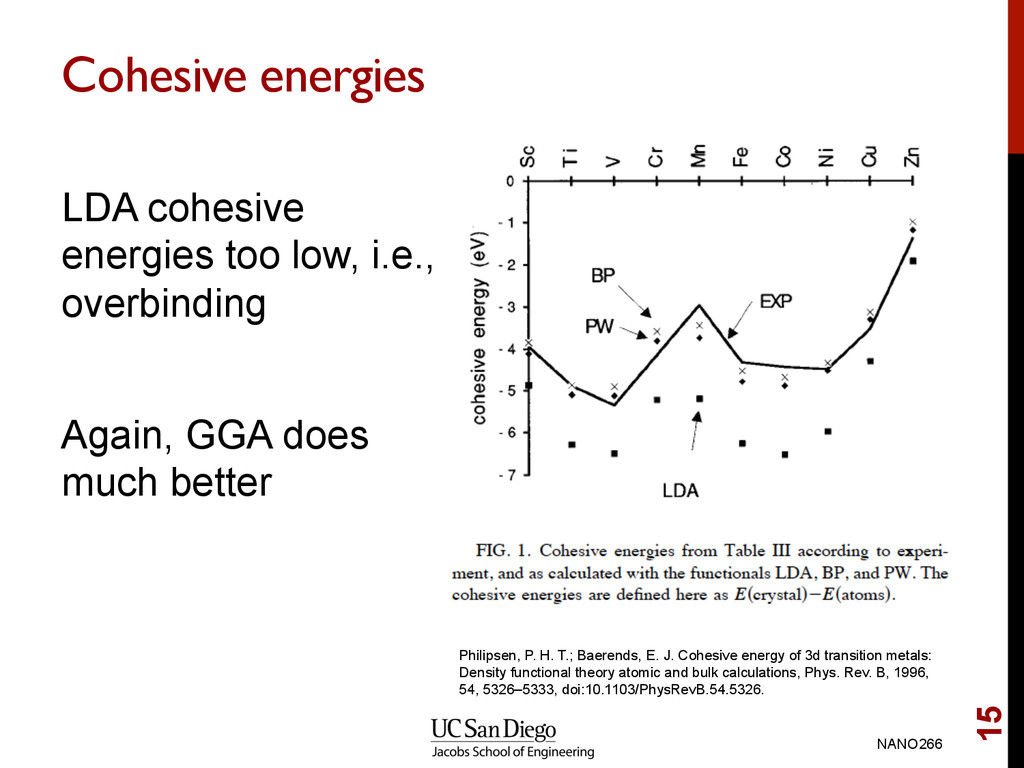
GGA does much better NANO266 15 Philipsen, P. H. T.; Baerends, E. J. Cohesive energy of 3d transition metals: Density functional theory atomic and bulk calculations, Phys. Rev. B, 1996, 54, 5326–5333, doi:10.1103/PhysRevB.54.5326.
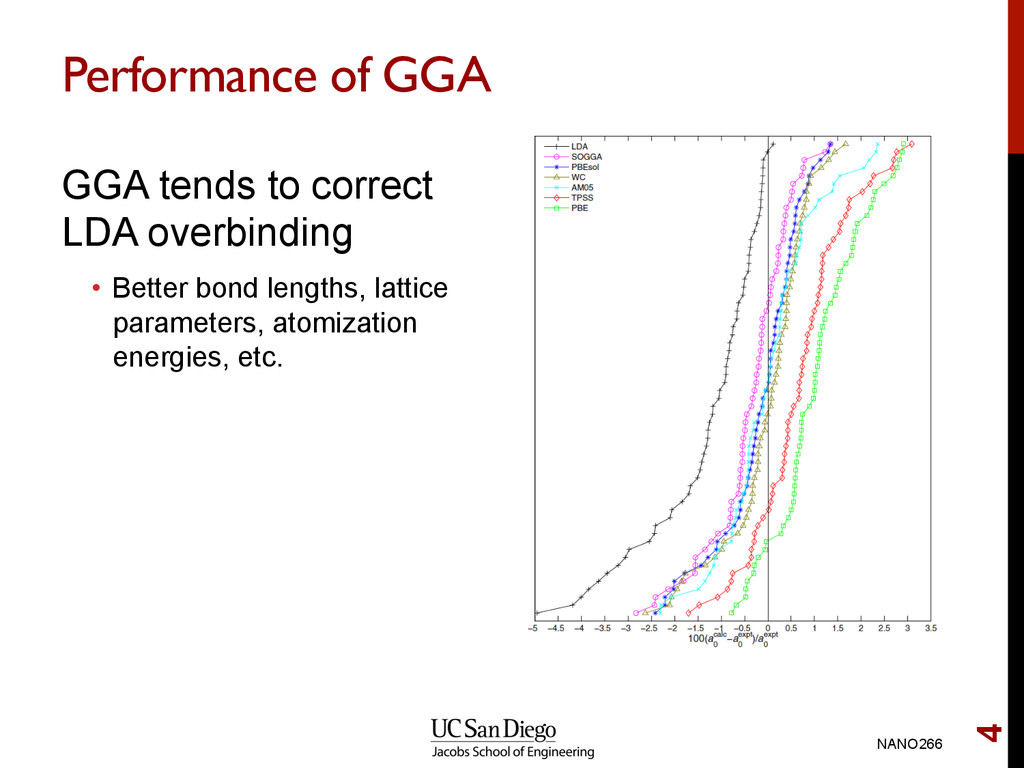
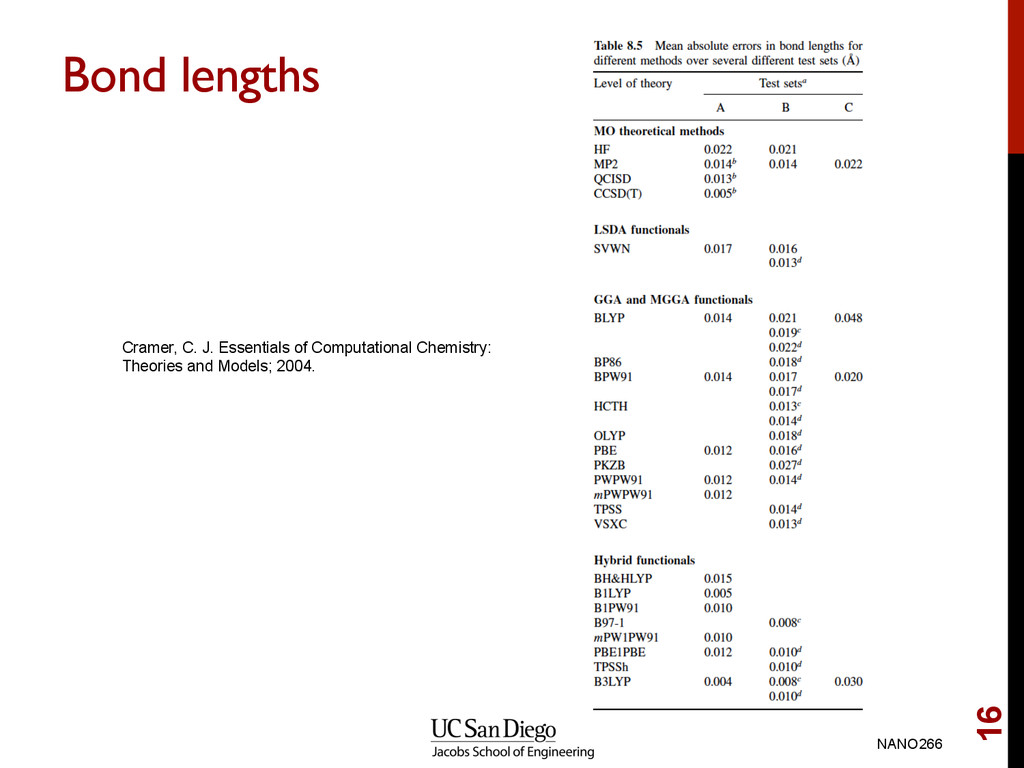
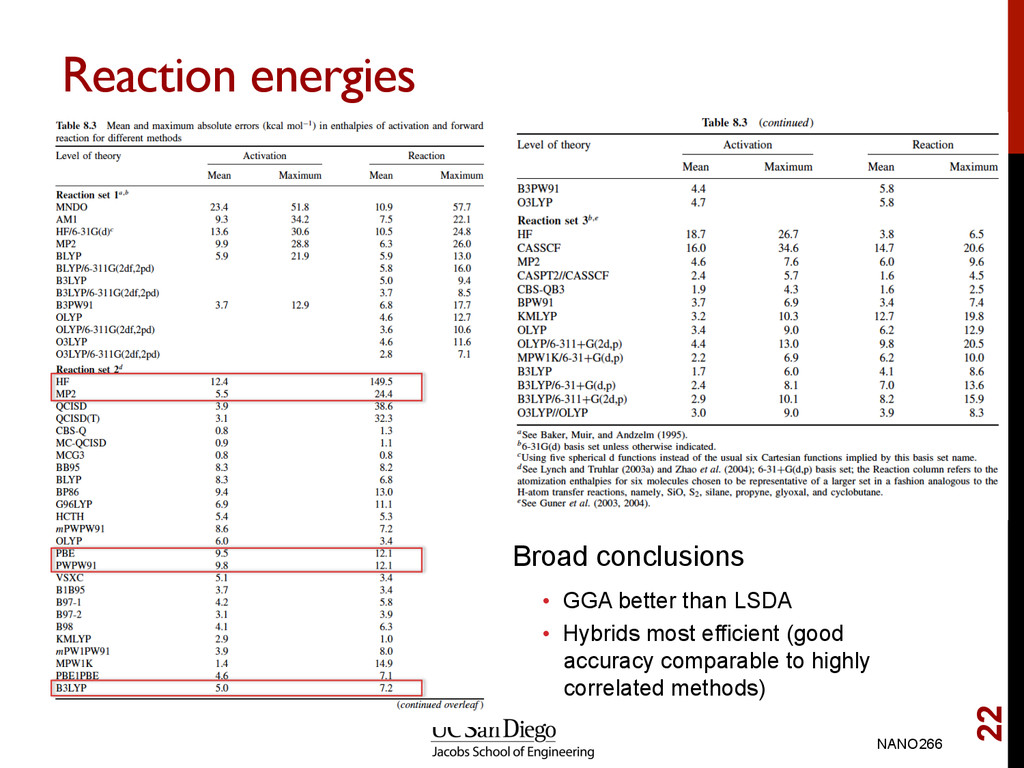
lengths, lattice parameters and overbinds GGA error is smaller, but less systematic. Error in GGA < 1% in many cases Conclusion • Very little reason to choose LDA over GGA since computational cost are similar Note: In all cases, we assume that LDA and GGA refers to spin-polarized versions. NANO266 17
Ry Bcc V : -1894.7125 Ry Cohesive energy = 0.638 Ry (0.03% of total E) Fcc/bcc difference = 0.02 Ry (0.001% of total E) Mixing energies ~ 10-6 fraction of total E NANO266 18 Ref: MIT 3.320 Lectures on Atomistic Modeling of Materials
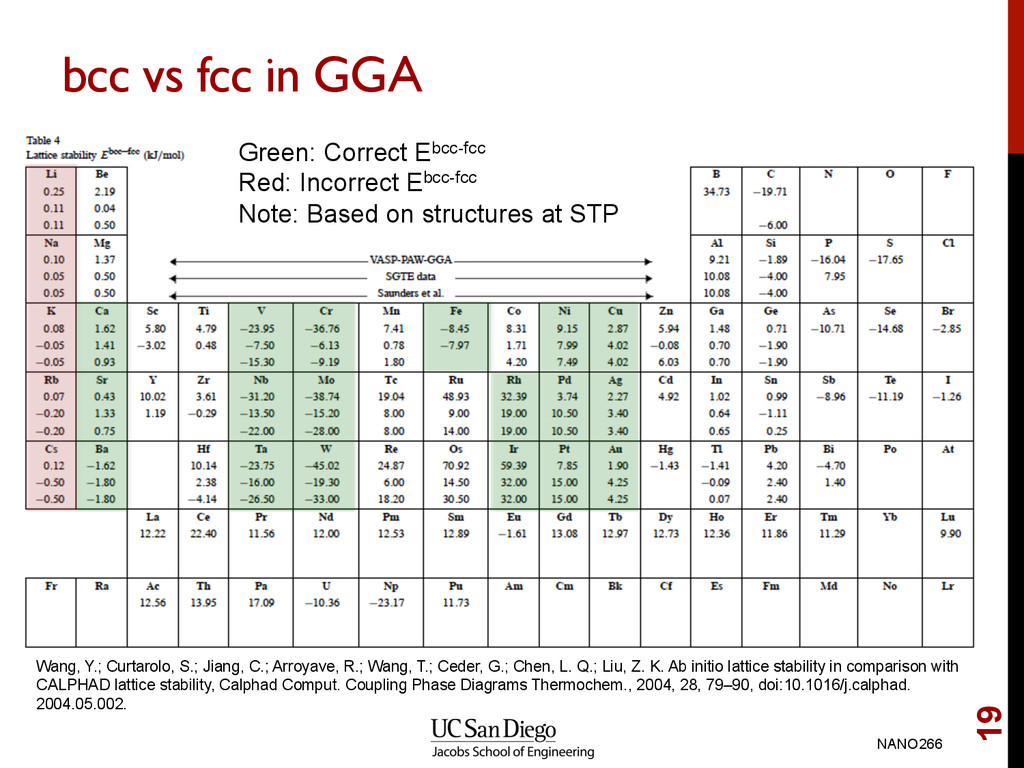
Red: Incorrect Ebcc-fcc Note: Based on structures at STP Wang, Y.; Curtarolo, S.; Jiang, C.; Arroyave, R.; Wang, T.; Ceder, G.; Chen, L. Q.; Liu, Z. K. Ab initio lattice stability in comparison with CALPHAD lattice stability, Calphad Comput. Coupling Phase Diagrams Thermochem., 2004, 28, 79–90, doi:10.1016/j.calphad. 2004.05.002.
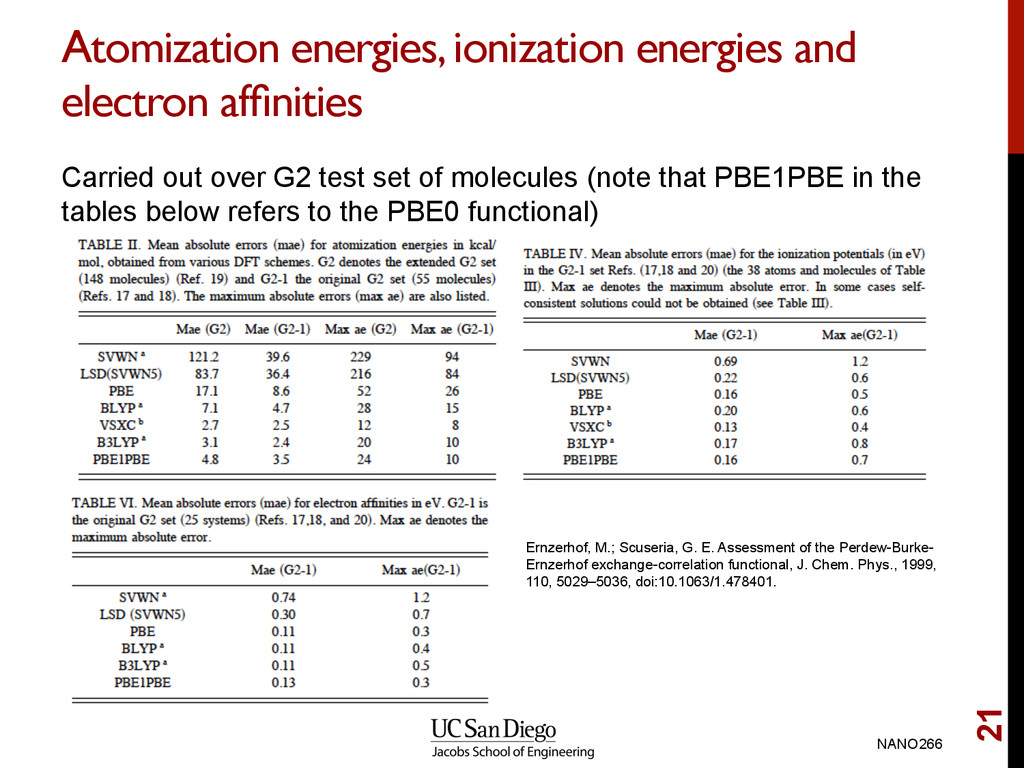
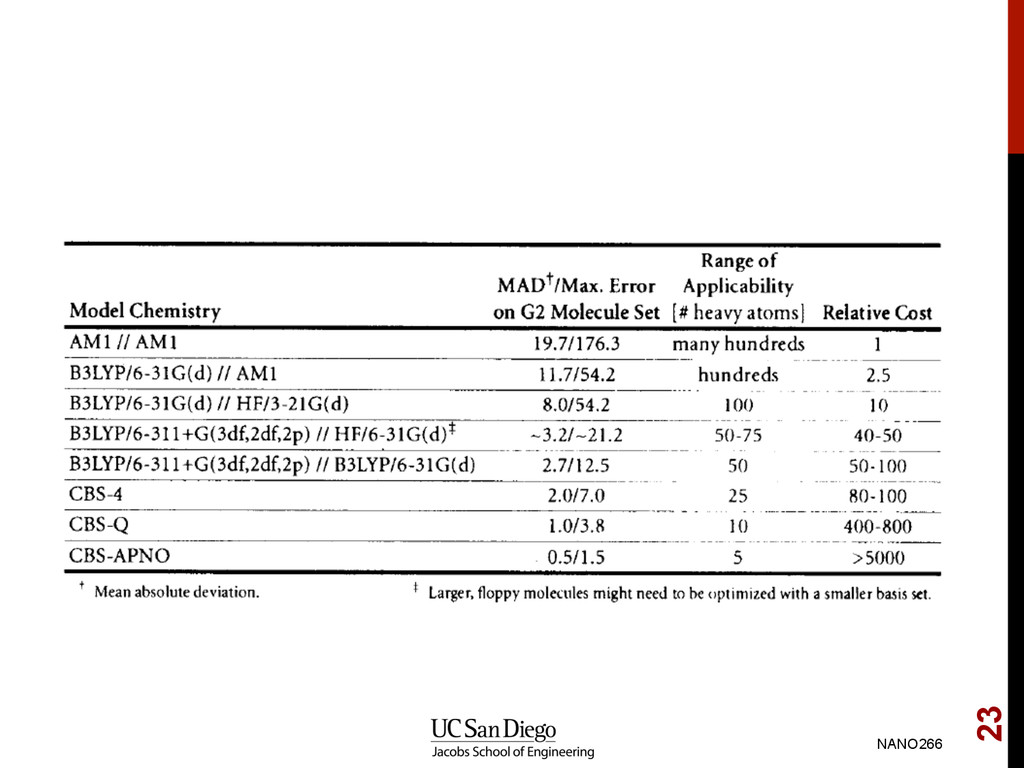
G2 test set of molecules (note that PBE1PBE in the tables below refers to the PBE0 functional) NANO266 21 Ernzerhof, M.; Scuseria, G. E. Assessment of the Perdew-Burke- Ernzerhof exchange-correlation functional, J. Chem. Phys., 1999, 110, 5029–5036, doi:10.1063/1.478401.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}