feature extraction (FE) applied to bioinformatics analysis Yh. Taguchi Department of Physics, Chuo University, Tokyo, Japan Presentation ORCID (papers)
# of samples < # of variables = no results Supervised Learning: Over fitting. Statistical tests: no positive hits because of adjusted Pvalues considering multiple comparisons (i.e., if the number of variables is 10,000, P = 0.0001 can happen by accident). → new methodology is required.
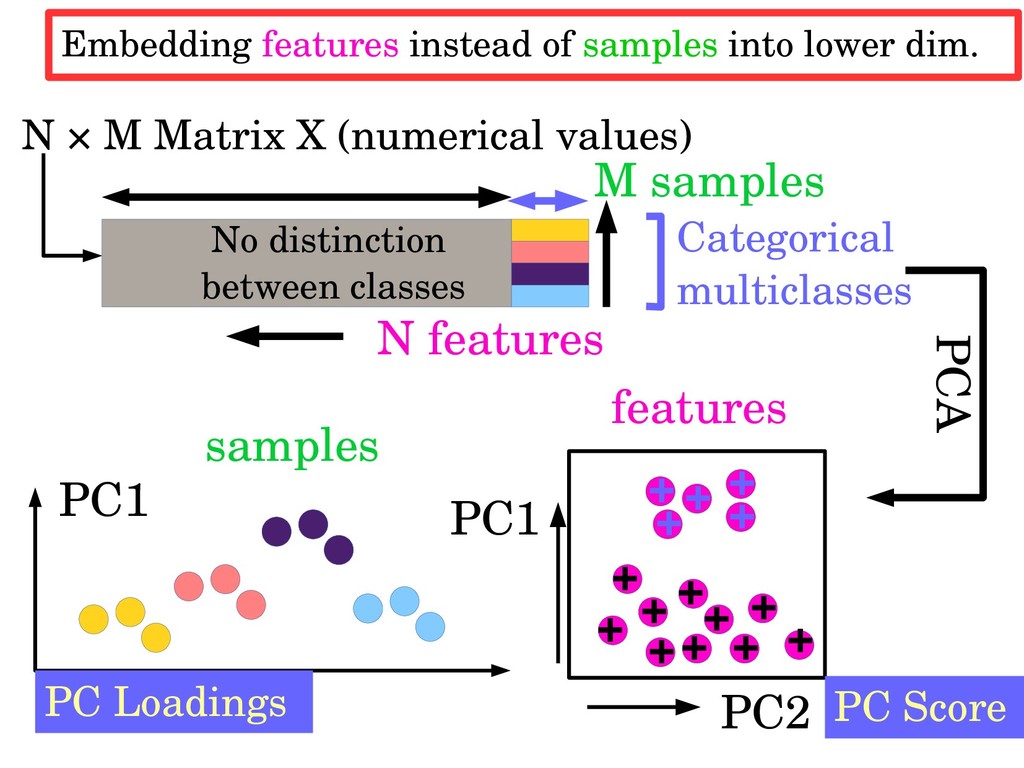
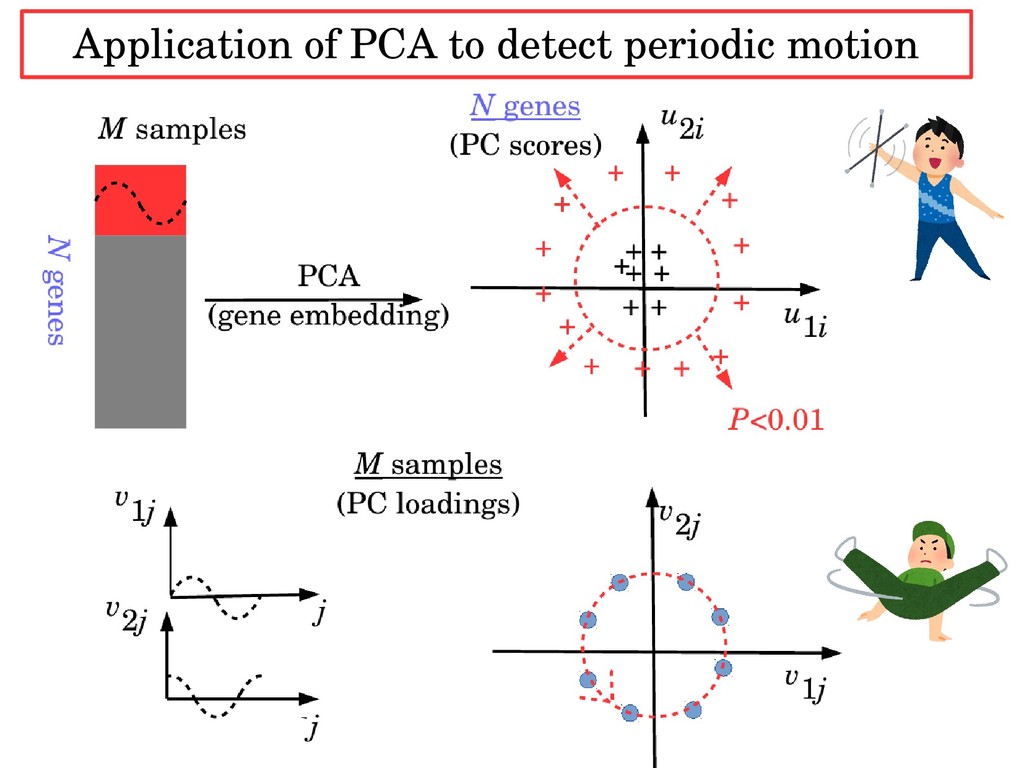
samples N × M Matrix X (numerical values) PC2 PC1 PC Score features + + + + + + + + + + + + + + + No distinction between classes Embedding features instead of samples into lower dim.
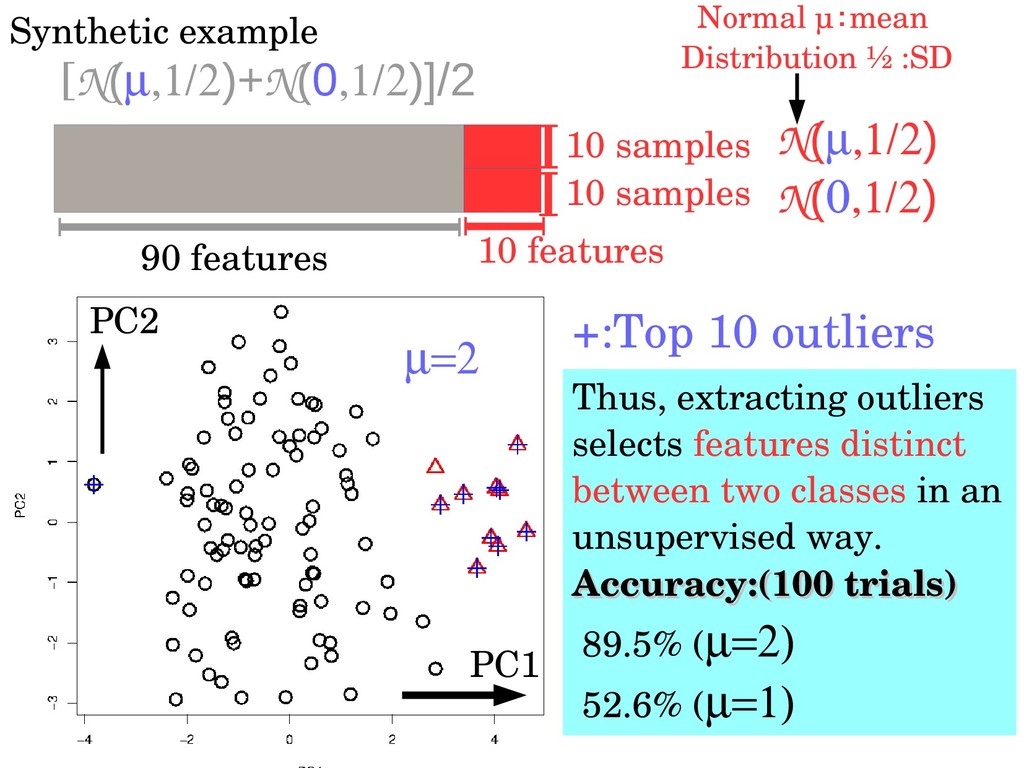
N(0) N() [N()+N(0)]/2 +:Top 10 outliers Thus, extracting outliers selects features distinct between two classes in an unsupervised way. Accuracy:(100 trials) Accuracy:(100 trials) 89.5% ( 52.6% ( PC1 PC2 Normal μ:mean Distribution ½ :SD
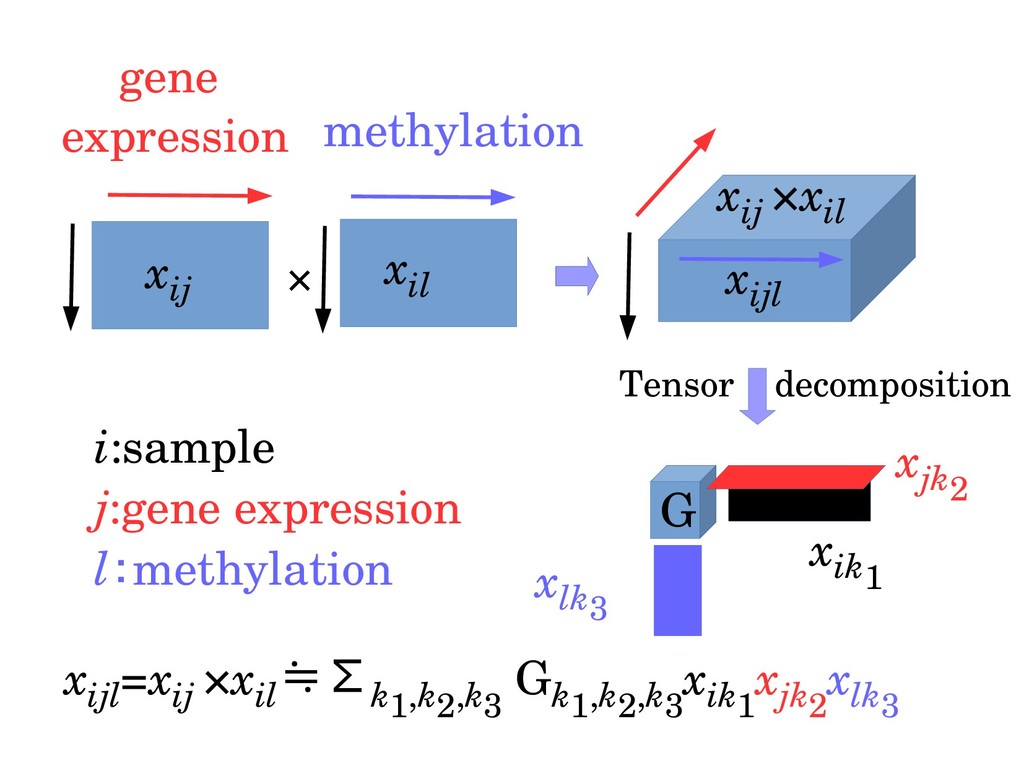
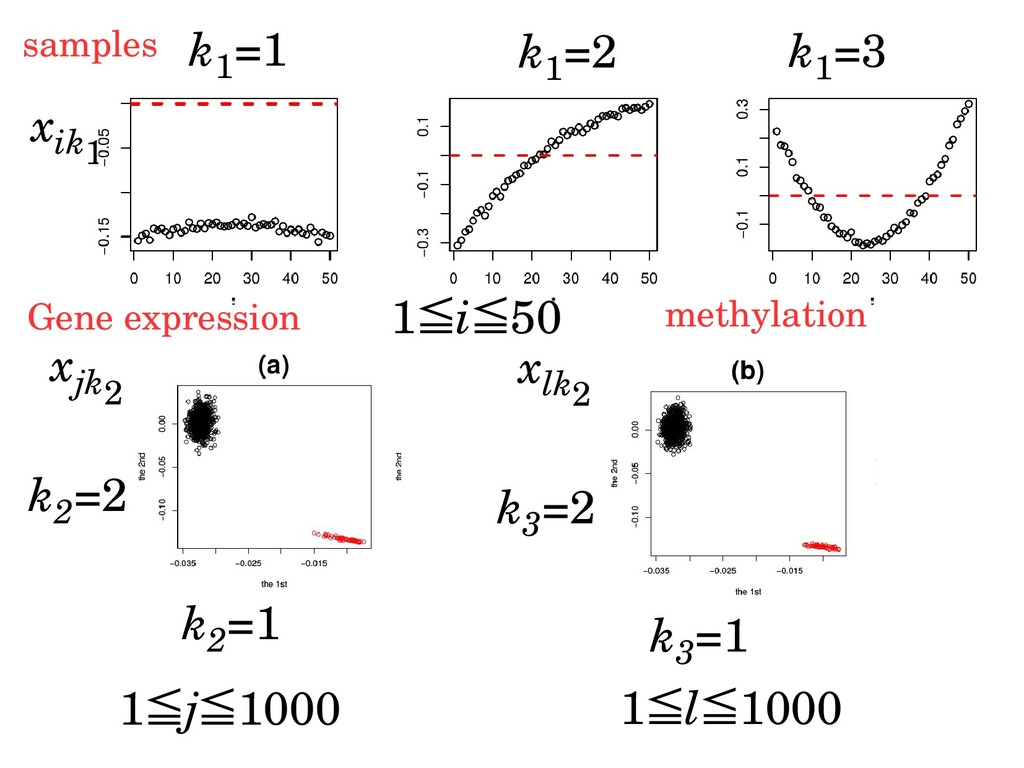
ijl Tensor decomposition G x ik1 x jk2 x lk3 x ijl =x ij ×x il ≒Σk1,k2,k3 G k1,k2,k3 x ik1 x jk2 x lk3 i:sample j:gene expression l:methylation gene expression methylation
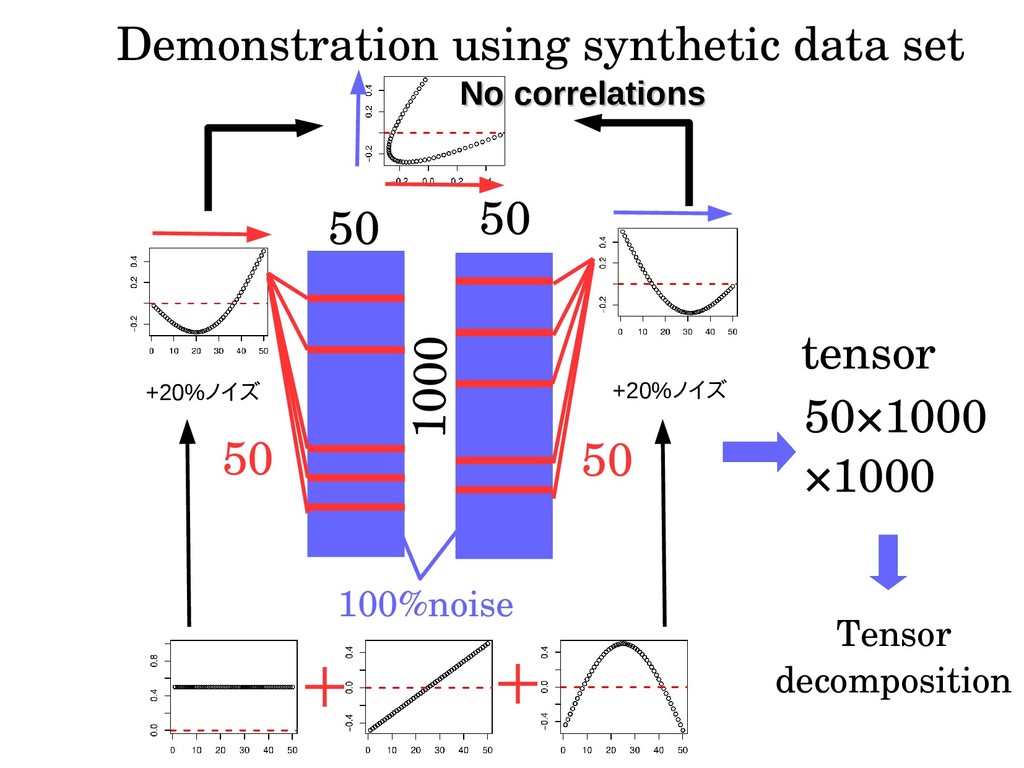
analysis tools ・No weights required to integrate multiple views ・Complete unsupervised learning (no model buildings using preknowledge) ・smaller computational resources because of linearity Disadvantages.... ・tendency to require more memories Solution:summing up Σi x ij ×x il results in j×l matrix that can be converted back (explains omitted)。 ・no shared feature or samples result in four mode.
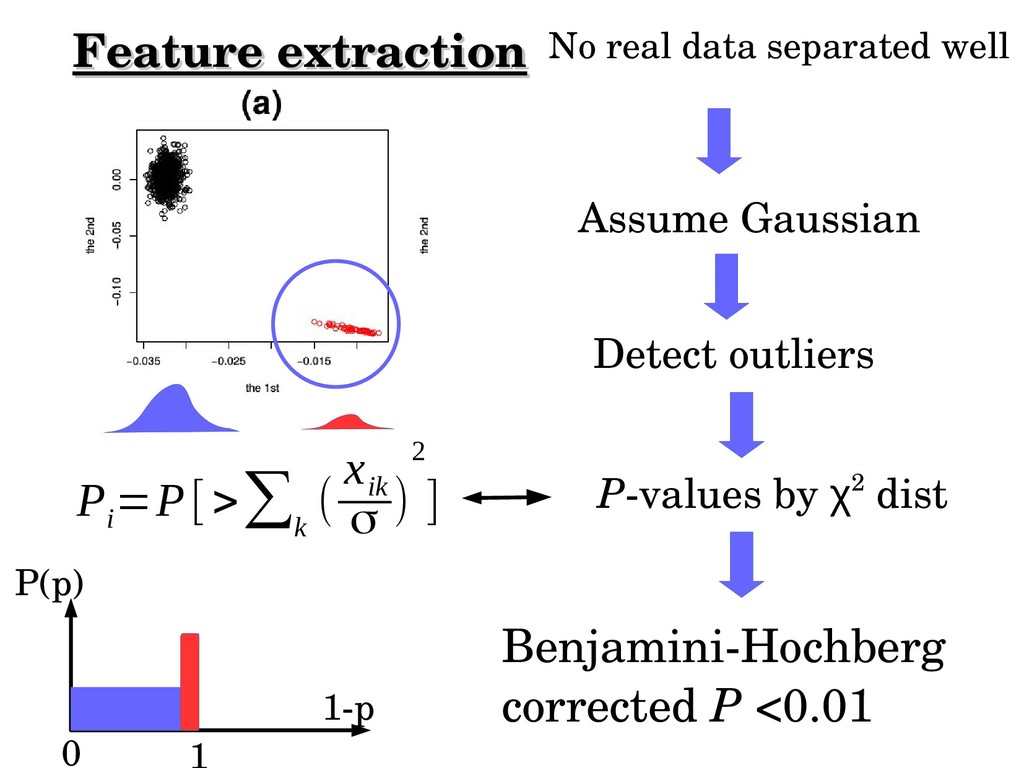
score obeys Gaussian distribution (Null hypothesis used also for probabilistic PCA) →Pvalues attributed to genes using χ2 distribution →Pvalues addjusted by Benjamini–Hochberg →Outlier genes: adjusted Pvalues<0.01 or 0.05
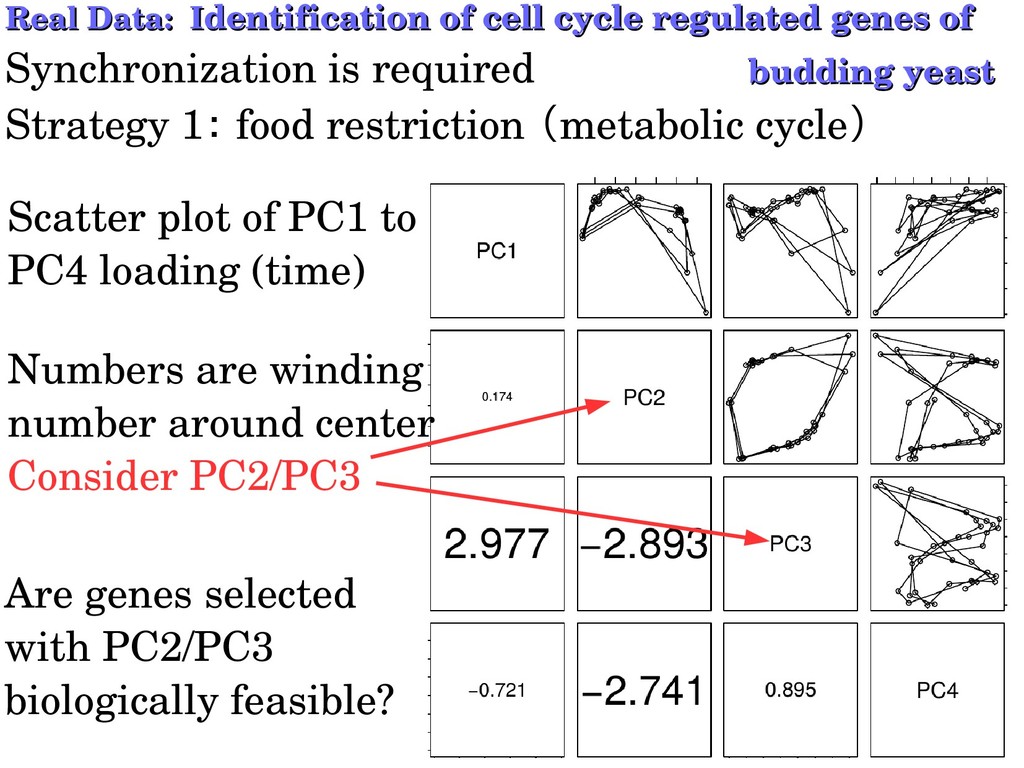
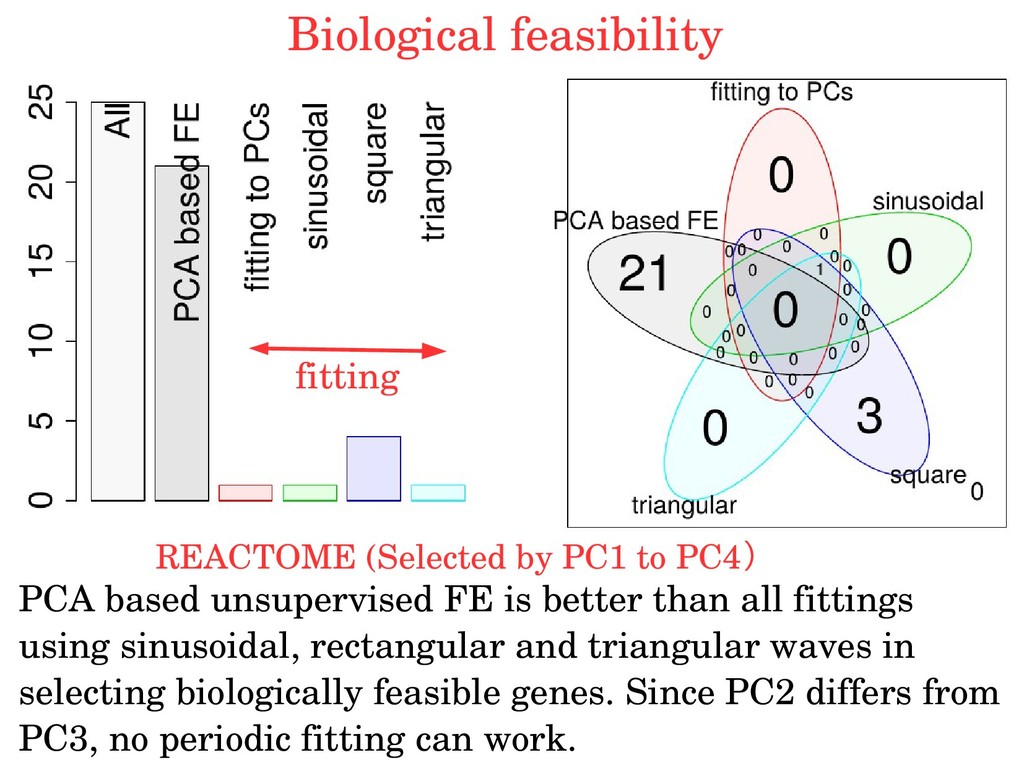
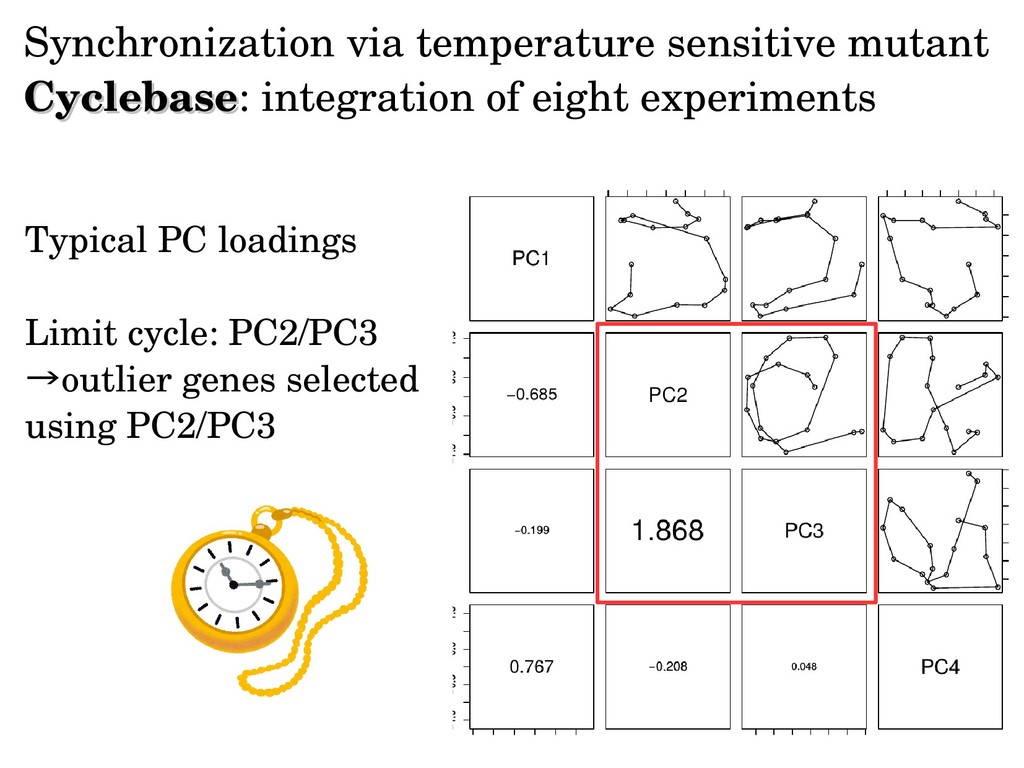
genes of dentification of cell cycle regulated genes of Synchronization is required budding yeast budding yeast Strategy 1: food restriction (metabolic cycle) Scatter plot of PC1 to PC4 loading (time) Numbers are winding number around center Consider PC2/PC3 Are genes selected with PC2/PC3 biologically feasible?
is better than all fittings using sinusoidal, rectangular and triangular waves in selecting biologically feasible genes. Since PC2 differs from PC3, no periodic fitting can work. fitting Biological feasibility
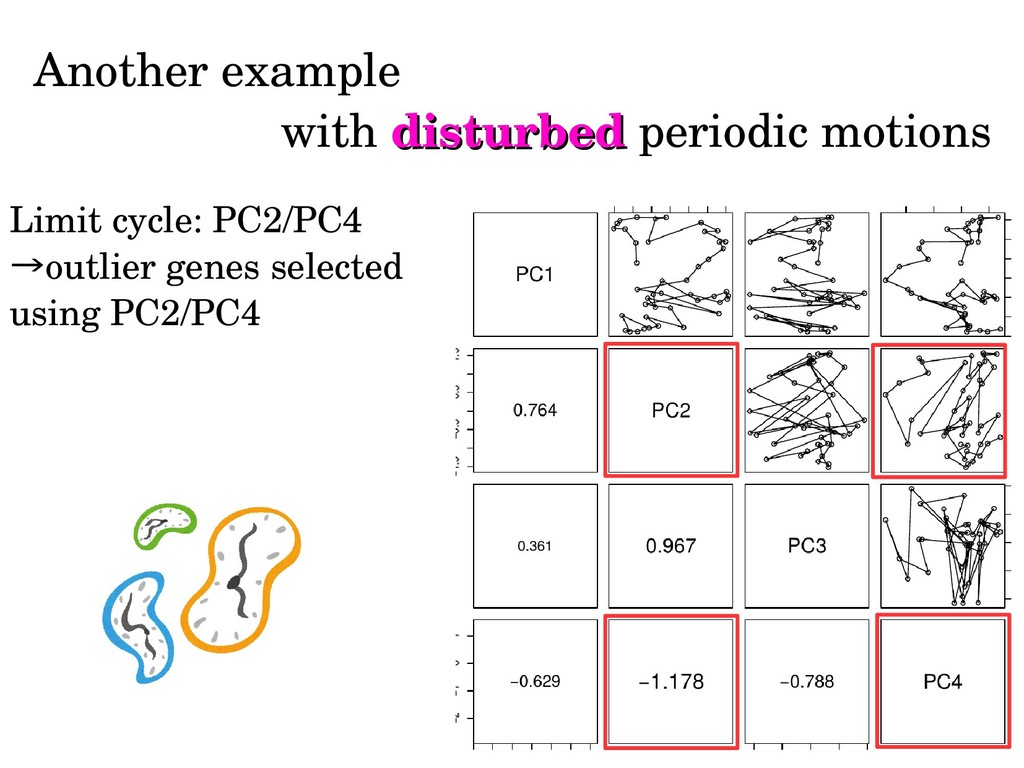
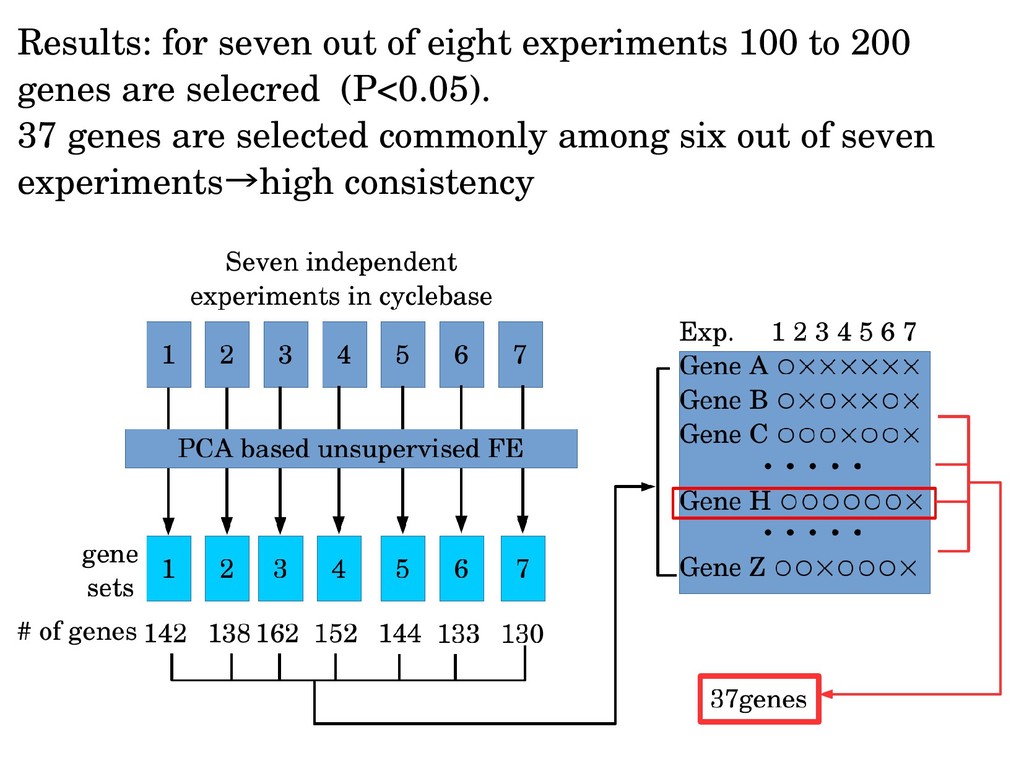
sinusoidal. Thus, sinusoidal regression might cause artifacts (But there will be no ways to assume true one a priori!) Limit cycle can be identified without functional forms or period. Biologically important three gene clusters can be identified in unsupervised way.
sample2 sample3 sample4 sample5 miRNA A group B group active active expression interaction x ij ×x il i:161samples, j:13393mRNA, l:755miRNA, (8 groups)
,k 3 ) 1≦k 1 k 2 k 3 5 ≦ k 1 :sample k 2 :mRNA k 3 :miRNA 1≦ k 2 5 ≦ Larger G Smaller G 1≦ k 3 2 ≦ x jk2 x lk3 assume Gaussian Detect outliers BenjaminiHochberg corrected P <0.01 Pvalues by χ2 dist 755miRNA中7miRNA 13393mRNA中427mRNA
genes SMID BREAST CANCER BREAST CANCER LUMINAL B DN SMID BREAST CANCER BREAST CANCER BASAL DN DOANE BREAST CANCER BREAST CANCER ESR1 UP SMID BREAST CANCER BREAST CANCER RELAPSE IN BONE DN SMID BREAST CANCER BREAST CANCER NORMAL LIKE UP FARMER BREAST CANCER BREAST CANCER BASAL VS LULMINAL BREAST CANCER BREAST CANCER UP SMID BREAST CANCER BREAST CANCER BASAL UP SMID BREAST CANCER BREAST CANCER LUMINAL B UP TURASHVILI BREAST DUCTAL CARCINOMA BREAST DUCTAL CARCINOMA VS DUCTAL NORMAL DN
of samples into lower dimension enable us to select variables of interest in an unsupervised manner. ・For the synthetic data set, variables distinct between two classes can be selected without labeling information. ・ For real data set (cell division cycle), PCA based unsupervised FE can identify gene with periodic motion without the knowledge of either functional form or period.
selection in multi view data, after applying tensor decomposition to a tensor generated by product of matrices, I propose to select features associated with BH corrected Pvalues <0.01 computed by χ2 dist assumed for a mode. ・ As for synthetic data set, apparently uncorrelated variables embedded into noised are decomposed to original orthogonal vectors after identifying correlated variables. ・As for muli omics data set, a few (a few %) intercorrelated and biologically reasonable miRNAs and mRNAs are identified among huge number of mRNAs and miRNAs
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}