Departamento de Qu´ ımica F´ ısica y Anal´ ıtica, Universidad de Oviedo http://web.uniovi.es/qcg/ Programa Interuniversitario de Doctorado en Qu´ ımica Te´ orica y Computacional Murcia, 21-01-2008 al 16-02-2008 c V. Lua˜ na, QTC Murcia 2008 (1)
The crystal lattice and crystal coordinates A periodic crystal can be described as a convolution of two elements: a periodic network of points (the lattice) and a molecular motif. + Molecular motif a b Lattice Cristal Convolution of two 1D functions: h(x) = f ◦ g(x) = Ω f(t) g(x − t) dt. Lattice: infinite but discrete network of points that exhibit translational symmetry. There exist a (non-unique) set of independent lattice vectors, {a, b, c}, such that all lattice points are connected by primitive translations: T ≡ {t = n1a + n2b + n3c | (n1, n2, n3) ∈ Z3} (1) c V. Lua˜ na, QTC Murcia 2008 (3)
definitions Unit cell: The {a, b, c} vectors define a parallelepipedon that re- produces the whole lattice network by means of primitive trans- lations. There is an inifinite number of ways for selecting a unit cell. Primitive cell: Any primitive cell contains exactly one lattice point and encloses the minimal volume that reproduces the whole crystal by translations alone. Centered cell: n primitive cells can be glued together to form a centered cell containing n lattice points. The use of a primitive cell means that the lattice is formed by primitive translations alone. The use of a centered cell means that the lattice contains fractional translations in addition to the primitive ones. Centered cells are used for convenience: showing better the symmetry of the crystal, evidencing the relationship of two structures, etc. c V. Lua˜ na, QTC Murcia 2008 (4)
b α γ β Crystal geometry Cell parameters: (a, b, c, α, β, γ). Director cosines: cos α = (b · c)/bc, cos β = (c · a)/ca, cos γ = (a · b)/ab. Cell volume: V = a · (b × c) = b · (c × a) = c · (a × b). Crystallographic coordinates: Any arbitrary point in the crystal is described by a vector: ri = xia + yib + zic = a b c xi yi zi = a ˜ xi , xi ∈ R3. (2) Main cell: Points in the main cell are such that 0 ≤ xi, yi, zi < 1. Any point in the crystal has an equivalent in the main cell, and it can be converted to it by a primitive translation. The crystal is an euclidean space described in terms of a non-orthonormal basis set. Most of the vector operations must be learned again. c V. Lua˜ na, QTC Murcia 2008 (5)
£ The scalar product (aka inner product) of two vectors is a measure- ment of the length of one of the vectors multiplied by the length of the perpendicular projection of the other on it. u · v = v · u = uv cos θ. (3) Vector’s norm: u = u = √ u · u. For two perpendicular vectors u · v = 0. The vector product of two vectors, u × v, is another vector such that: • it is perpendicular to both vector factors; • the length is equal to the area of the parallelogram defined by the vector factors, u × v = uv sin θ; • the orientation is given by the right hand rule (the thumb points as the vector product when the rest of fingers move from the first to the second factor). For any vector: u × u = 0. ¡ ¢ ¡¤£ ¢ ¢¥£ ¡ ¦ Mixed triple product (aka box product): u · (v × w) = v · (w × u) = w · (u × v) gives the volume of the parallelepipedon defined by the three vectors. c V. Lua˜ na, QTC Murcia 2008 (6)
using the crystallographic coordinates Vector addition: ri ± rj = a ˜ (xi ± xj ) = a b c xi ± xj yi ± yj zi ± zj . (4) Scalar product: ri · rj = (a ˜ xi )T(a ˜ xj ) = xT i a ˜ T a ˜ xj = xT i G xj = xi yi zi G xj yj zj , (5) where G = a ˜ Ta ˜ = a b c a b c = a · a a · b a · c b · a b · b b · c c · a c · b c · c = a2 ab cos γ ac cos β ab cos γ b2 bc cos α ac cos β bc cos α c2 (6) is the metrical matrix (aka metrical tensor) of the lattice. For the orthonormal {i, j, k} basis G = 1. The determinant of the metrical tensor is the square of the cell volume: V 2 = det G = G =⇒ V = abc 1 − cos2 α − cos2 β − cos2 γ + 2 cos α cos β cos γ (7) Vector products can be simplified by the introduction of the reciprocal lattice construction. c V. Lua˜ na, QTC Murcia 2008 (7)
cell and the vector product The reciprocal of the (a, b, c) cell is formed by the (a , b , c ) vectors, defined is such a way that the reciprocal of a cell vector has an inverse length and it is perpendicular to the other two vectors of the cell. In other words: (a ˜ )T a ˜ = s1 =⇒ a b c a b c = s 1 0 0 0 1 0 0 0 1 (8) where s is an adimensional factor. In solid state physics is customary to choose s = 2π, whereas the crystallographical and mathematical convention is s = 1. We will use s = 1 for reasons that will be later explained. The reciprocal lattice definition is satisfied by choosing: a = b × c V , b = c × a V , c = a × b V . (9) Direct and reciprocal cell have inverse volumes: V V = 1 (V = a · (b × c )). Additionally a = b × c V , b = c × a V , c = a × b V . (10) Therefore, our original cell (aka direct cell) is the reciprocal of the reciprocal cell: (a ˜ ) = a ˜ . c V. Lua˜ na, QTC Murcia 2008 (8)
parameters: (a , b , c , α , β , γ ). These parameters can be obtained from the direct cell lenghts and angles as a = bc sin α V , b = ca sin β V , c = ab sin γ V , (11) cos α = cos β cos γ− cos α sen β sen γ , cos β = cos γ cos α− cos β sen γ sen α , cos γ = cos α cos β− cos γ sen α sen β . (12) Notice that if a, b, c [=] ˚ A then a , b , c [=] ˚ A−1. Vector product (of two direct cell vectors): ri × rj = (xia + yib + zic) × (xja + yjb + zjc) = yi zi yj zj (b × c) V a − xi zi xj zj (c × a) V b + xi yi xj yj (a × b) V c = V a b c xi yi zi xj yj zj (13) Direct and reciprocal vectors. The next convention will be used to distinguish among them: r = a ˜ x, h = a ˜ h, κ = 2πh = a ˜ κ. (14) Thus κ recovers the solid state convention of incorporating the 2π factor. c V. Lua˜ na, QTC Murcia 2008 (9)
on either the direct or reciprocal cell requires a metrical matrix, but the mixed product of a vector from one space and a vector from the other does not require it: ri · rj = xT i G xj , hi · hj = hT i G xj , h · r = hT x, κ · r = κT x, (15) where G = G−1 = (a ˜ )Ta ˜ = (a )2 a b cos γ a c cos β a b cos γ (b )2 b c cos α a c cos β b c cos α (c )2 . (16) Vector product ri × rj = V a b c xi yi zi xj yj zj , hi × hj = V a b c hi ki li hj kj lj , κi × κj = V a b c κxi κyi κzi κxj κyj κzj , (17) Mixed triple product ri · (rj × rk) = V xi yi zi xj yj zj xk yk zk , hi · (hj × hk) = V hi ki li hj kj lj hk kk lk . (18) c V. Lua˜ na, QTC Murcia 2008 (10)
the lattice vectors Linear transformations: They imply a change in the orientation, length, or both of the basis vectors. The position vectors do not change with the axis transformation. a b c = a b c p11 p12 p13 p21 p22 p23 p31 p32 p33 =⇒ r = a ˜ x = a ˜ P x = a ˜ x = r (19) The transformation can be interpreted as operating on the axes or on the coordinates: a ˜ = a ˜ P ⇐⇒ x = P x ⇐⇒ x = P−1x ⇐⇒ a ˜ = a ˜ P−1. (20) Special case: an orthogonal transformation, P−1 = PT, represents a proper (if Det P > 0) or improper (if Det P < 0) rotation of the cell axes. Origin shift: The initial origin, O, is moved to O by a translation p = a ˜ p. This modifies all position vectors: r −→ r = r − p, or a ˜ x = a ˜ (x − p). Affine transformation: General operations have both, linear and shift terms: (P|p)x = P x + p. c V. Lua˜ na, QTC Murcia 2008 (11)
the reciprocal space The reciprocal lattice. Translational symmetry in the direct space also implies translational symmetry in the reciprocal space. If t is a primitive translation: ∀ r∈R3 r + t = r =⇒ eih·r = eih·(r+t) = eih·reih·t =⇒ eih·t = 1. (21) The network of lattice points in the direct space is transformed into a network of reciprocal lattice points through the condition eih·t = 1. Crystalographic planes. In Miller’s notation, indices (h, k, l) define a plane intersecting the a, b and c axes at a distance a/h, b/k and c/l, respectively, from the origin. (0,0,1) (1,1,1) (1,0,2) (1,−1,2) (0,2,0) Common notations: [uvw] a direction in the crystal; <uvw> a set of directions equivalent by symmetry; (hkl) Miller indices of a plane; {hkl} a set of planes equivalent by symmetry. c V. Lua˜ na, QTC Murcia 2008 (12)
b c c/l b/k v 1 v 2 H O Normal vector to a crystallographic plane: Vectors v1 = b/k − a/h and v2 = c/l − b/k lie in (h, k, l) plane. Their vectorial product: h = f(v1 × v2) = ... = fV hkl (ha + kb + lc ) (22) is perpendicular to the plane. Using f = hkl/V as a normalization factor, we see that the reciprocal lattice vector h = a ˜ h = ha +kb +lc is the normal to all the planes (λh, λk, λl), where λ ∈ R is a scale factor. Planes in direct space are associated to directions in the reciprocal space, and viceversa. Both spaces form a dual. Special hexagonal plane indexing: • Plane (hkil), where i is the reciprocal of the fractional intercept of the a3 axis. The three first indices are not independent, but h + k = −i. • Direction [uvtw], related to the usual [UV W] notation by: U = u−t, V = v − t, W = w. a 1 a 2 a 3 c (1100) − (1011) − c V. Lua˜ na, QTC Murcia 2008 (13)
crystal systems and the fourteen Bravais lattices The 7 crystal systems appear as a consequence of imposing symmetry conditions upon the lattice vectors. The consideration of primitive or centered cells on the crystal systems produces 14 different cell types known as Bravais lattices. System Symmetry Cell parameters Bravais latt. Cubic Four C3(3) axes a = b = c, α = β = γ = 90◦ P, I, F Hexagonal a C6(6) or S3(¯ 6) axis a = b, α = β = 90◦, γ = 120◦ P Trigonal (H) a C3(3) or S6(¯ 3) axis a = b, α = β = 90◦, γ = 120◦ R Trigonal (R) a C3(3) or S6(¯ 3) axis a = b = c, α = β = γ R Tetragonal a C4(4) or S4(¯ 4) axis a = b, α = β = γ = 90◦ P, I Orthorhombic three ⊥ C2(2) or S2(¯ 2) axes α = β = γ = 90◦ P, I, F. C Monoclinic a C2(2) or S2(¯ 2) axis α = β = 90◦ or α = γ = 90◦ P, B or C Triclinic E(1) or i(¯ 1) none P Trigonal or rhombohedral: The true R cell is primitive, but the R cell with hexagonal axes contains three lattice points and it is centered on (2/3, 1/3, 1/3) (obverse) or (1/3, 2/3, 1/3) (reverse). c V. Lua˜ na, QTC Murcia 2008 (15)
P: primitive; I: centered in the middle point (Inner); F: centered on each cell face (Faces); A (B or C): centered of the faces perpendicular to the a lattice vector (A) or to the b (B) or c (C); R: rhombohedral (primitive only when rhombohedral axes are used). Cubic P (sc) Cubic I (bcc) Cubic F (fcc) Hex. P Trigonal R Tetrag. P Tetrag. I Ortho. P Ortho. I Ortho. F Ortho. C Mono. P Mono. B Tric. P c V. Lua˜ na, QTC Murcia 2008 (16)
cell can be converted to a primitive form: F: I: C: aP (a + b)/2 (a + b − c)/2 (a − b)/2 bP (b + c)/2 (−a + b + c)/2 (a + b)/2 cP (c + a)/2 (a − b + c)/2 c c P a c b a P b P a b c c P bP a P c P c a b P a P b = c V. Lua˜ na, QTC Murcia 2008 (17)
case of the R cell: Using hexagonal axes, the cell can be centered at: • (0, 0, 0), (2/3, 1/3, 1/3), (1/3, 2/3, 2/3) (Obverse set- ting, modern, ITC-A). • (0, 0, 0), (1/3, 2/3, 1/3), (2/3, 1/3, 2/3) (Reverse set- ting, classical, IT1). A rhombohedral cell for the obverse setting: • 3aR = 2aH + bH + cH , • 3bR = −aH + bH + cH , • 3cR = −aH − 2bH + cH . Up to six different fully different hexagonal-rhombohedral arrangements can be described. The cell described with hexagonal axes contains 3 primi- tive R cells. c V. Lua˜ na, QTC Murcia 2008 (18)
b, c, 90, β, 90) V = abc sin β, x = [(ax)2 + (by)2 + (cz)2 + 2(ax)(cz) cos β]1/2, (33) (1/a sin β, 1/b, 1/c sin β, 90, β + π, 90), (34) h = [(ha )2 + (kb )2 − 2hla c cos β + (lc )2]1/2. (35) Triclinic: V = abc[1 − cos2 α − cos2 β − cos2 γ + 2 cos α cos β cos γ]1/2, V = V −1, (36) x = (ax)2 + (by)2 + (cz)2 + 2(ax)(by) cos γ + 2(by)(cz) cos α + 2(cz)(ax) cos β 1/2 , (37) (a , b , c , α , β , γ ), (38) a = bc V sin α, sin α = V a b c , cos α = cos β cos γ − cos α sin β sin γ , (39) b = ca V sin β, sin β = V b c a , cos β = cos γ cos α − cos β sin γ sin α , (40) c = ab V sin γ, sin γ = V c a b , cos γ = cos α cos β − cos γ sin α sin β , (41) h = (a h)2 + (b k)2 + (c l)2 + 2(a h)(b k) cos γ + 2(b k)(c l) cos α + 2(c l)(a h) cos β 1/2 . (42) c V. Lua˜ na, QTC Murcia 2008 (20)
(WS) cell Crystalographic cells are parallelepipedic by convenience, but they could have arbitrary forms and still reproduce the crystal through the symmetry translations. Wigner-Seitz lattices are conceptually quite important. A WS cell about a lattice point is the crystal region closer to that point than to any other lattice point. To form the WS cell, a lattice point is connected by lines to its lattice neighbors, and each line is divided into two halves by mean of a perpendicular plane. The WS cell is the smallest polyhedron that is interior to all the planes. bcc fcc c V. Lua˜ na, QTC Murcia 2008 (21)
Molecular symmetry is usually described on Sch¨ onflies’ scheme, but the Hermann-Mauguin (aka IUCr) scheme is preferred on crystals. The main difference lies in the selection of improper rotations: Sch¨ onflies uses rotation-reflection operations, whereas Hermann-Mauguin is based on rotation-inversion operations. Both schemes are equivalent but not the same. Hermann-Mauguin: n (= 1, 2, ...) is a rotation axis of order n (1 is the identity); ¯ n is a rotation- inversion axis of order n (¯ 1 is the inversion itself); m is a reflecting plane; n|m means a mirror plane perpendicular to a n axis; and nm indicates n mirror planes that contain a n rotation axis. H-M (IUCr) 1 n m n|m nm ¯ 1 ¯ 2 ¯ 3 ¯ 4 ¯ 5 ¯ 6 . . . Sch¨ onflies E Cn σ Cn y σh Cn y nσv i σ S6 S4 S10 S3 . . . 3 1 2 3 6 4 5 1 6 3 5 2 6 4 S 3 1 3 5 4 2 6 1 S 6 5 3 6 2 4 c V. Lua˜ na, QTC Murcia 2008 (22)
groups Molecules can have symmetry axes of any order. Translational invariance, however, excludes proper axes of order 5 and ≥ 7. Only 32 point groups are thus compatible with the 3D crystal lattices. System Essential N◦ Notation: Sch¨ onflies (HM-IUCr) Cubic 4 × 3 (C3) 4 T (23), Td(¯ 43m), Th(m3), O(432), Oh(m3m). Tetrag. 4 (C4) 7 S4(¯ 4), C4(4), C4v(4mm), C4h(4|m), D2d(¯ 42m), D4(422), D4h(4|mmm). Hexag. 6 (C6) 6 C6(6), C6v(6mm), C6h(6|m), D6(622), D6h(6|mmm), S6(¯ 3). 3 (C3) 6 C3(3), C3v(3m), C3h(¯ 6), D3(32), D3h(¯ 6m2), D3d(¯ 3m). Ortho. 222 or mm 3 D2(222), D2h(mmm), C2v(2mm). Monoc. 2 or m 3 C2(2), Cs(m), C2h(2|m). Tric. – 2 C1(1), Ci(¯ 1). The molecular motif can include C5 axes (for instance C60 or some bacteriophage virii) but the forbidden axes do not belong to the crystal. Quasicrystals, lacking true translational symmetry, use to show pentagonal and heptagonal local symmetry. c V. Lua˜ na, QTC Murcia 2008 (23)
Seitz operators Crystal symmetry operations are affine transformations, including rotational and translational parts: r i = { ˆ R|ˆ t} ri = R xi + t = r11 r12 r13 r21 r22 r23 r31 r32 r33 xi yi zi + tx ty tz . (43) • Pure rotation: { ˆ R|ˆ 0}. • Pure translation: {ˆ 1|ˆ t}. • Product of operations: { ˆ R|ˆ tR}{ ˆ S|ˆ tS} = { ˆ R ˆ S|ˆ tR + ˆ Rˆ tS}. • Unit operation (neutral): {ˆ 1|ˆ 0} =⇒ {ˆ 1|ˆ 0}{ ˆ R|ˆ tR} = { ˆ R|ˆ tR}{ˆ 1|ˆ 0} = { ˆ R|ˆ tR}. • Inverse operation: { ˆ R|ˆ tR}−1 = { ˆ R−1| − ˆ R−1ˆ tR}. • Null operation: {ˆ 0|ˆ 0} =⇒ {ˆ 0|ˆ 0}{ ˆ R|ˆ tR} = { ˆ R|ˆ tR}{ˆ 0|ˆ 0} = {ˆ 0|ˆ 0}. The set of symmetry operations in a crystal forms a mathematical group: the product of any two operations is another symmetry operation; there exists a single unit operation; and any operation has an inverse that also belongs to the group. This space group of the crystal contains inifinite operations, because the rotational part can be arbitrarily combined with any primitive translation (symmorphic operations). c V. Lua˜ na, QTC Murcia 2008 (24)
The space group can also contain operations in which a fractionary translation is combined with a non null rotational part. • Screw axes (aka rotation-translation axes). A nk operation is a rotation of 2π/n followed by a translation of k/n parts of the cell lenght along the rotation axis. Ex: 32[0, 0, 1] = {3[0, 0, 1]|τ(0, 0, 2/3)}. Possible cases: 21, 31, 32, 41, 42, 43, 61, 62, 63, 64 y 65. • Glide planes: Composed of the reflection in a plane plus a fractional translation. The translation can be: axial (a, b, or c): the translation (a/2, b/2 or c/2) is contained within the plane (ab, bc or ac). Ex: {m[100]|τ(0, 1/2, 0)}. diagonal (n): glides along a diagonal face ((a + b)/2, (b + c)/2 or (c + a)/2). Ex: {m[001]|τ(1/2, 1/2, 0)}. diamond (d): translation along a (a ± b)/4, (b ± c)/4, (c ± a)/4, or (a + b + c)/4 cubic direction. c V. Lua˜ na, QTC Murcia 2008 (25)
Space group (G): Formed by the infinite symmetry operations of the crystal. Translation group (T ): The infinite set of primitive translations also forms a group. Factor group (G/T ): This quotient group removes from G all operations containing translations that involve displacements of one cell length or more on any direction. Centering translations are left. The result is a finite group. Most references to the space group in the literature correspond to G/T rather than to G. Point group (Gp): Further collapse of all translations to 0 produces the associated point group, that describes exclusively the rotational symmetry of the crystal. Laue class: In the absence of anomalous scattering the diffraction pattern of a crystal shows inversion invariance even if the crystal lacks it (Friedel’s law). The Laue class, which adds the inversion to the point group, is the symmetry of the diffraction pattern. The Bravais centering added to the Laue class forms the Patterson symmetry. Gp and G/T dimension: Gp P I,A,B,C R (R axes) R (H axes) F Size h h 2h h 3h 4h Example: The cubic m¯ 3m (Oh, h = 48) point group gives rise to three different space groups: Pm¯ 3m, Im¯ 3m and Fm¯ 3m, of dimensions 48, 96 and 192, respectively, c V. Lua˜ na, QTC Murcia 2008 (26)
space groups By 1891 E. S. Fedorov and A. M. Sch¨ onflies, working independently, completed the list of the 230 crystallographic space groups. 73 of these are symmorphic groups that come from the combination of the 14 Bravais lattices and the 32 crystallographic point groups. The 157 remaining non-symmorphic groups include glide translations and screw rotations. System #SG SG Bravais Point groups Laue class Triclin. 2 1–2 P 1, ¯ 1 ¯ 1 Monoc. 13 3–15 P, C 2, m, 2|m 2|m Ortho. 59 16–74 P, C, I, F 222, mm2, mmm mmm Tetrag. 68 75–142 P, I 4, ¯ 4, 4|m, 422, 4mm, ¯ 42m, 4|mmm 4|m, 4|mmm Trig. 25 143–167 P, R 3, ¯ 3, 32, 3m, ¯ 3m ¯ 3, ¯ 3m Hexag. 27 168–194 P 6, ¯ 6, 6|m, 622, 6mm, ¯ 62m, 6|mmm 6|m, 6|mmm Cubic 36 195–230 P, I, F 23, m¯ 3, 432, ¯ 43m, m¯ 3m m¯ 3, m¯ 3m The space groups and their main properties are described in the International Tables for Crystal- lography. The Bilbao Crystallographic Server (http://www.cryst.ehu.es) provides a high quality tool on Internet. c V. Lua˜ na, QTC Murcia 2008 (27)
Tables of Crystal- lography Posiciones de Wyckoff Extinciones sistematicas Vectores de centrado Sistema cristalino Grupo Espacial Grupo puntual c V. Lua˜ na, QTC Murcia 2008 (28)
The application of the factor group symmetry operations on the coordinates of an arbitrary point produces all the equivalent positions of that point. For instance, a generic (x, y, z) has 8 equivalent positions in the P42/m group (num. 84): (x, y, z) 42 − − − − − → (¯ y, x, z + 1/2) 42 − − − − − → (¯ x, ¯ y, z) 42 − − − − − → (y, ¯ x, z + 1/2) m m m m (x, y, ¯ z) 42 − − − − − → (¯ y, x, 1/2 − z) 42 − − − − − → (¯ x, ¯ y, ¯ z) 42 − − − − − → (y, ¯ x, 1/2 − z). (44) Position (x, y, z) is said to have a multiplicity of 8 in this group. The multiplicity of a general position is equal to the order nh of the G/T factor group. However, if a point is contained within one or more of the symmetry elements, several of the symmetry operations will produce the same image when applied to the point coordinates. The real multiplicity of the position will be reduced to a divisor of nh. For instance, in the same P42/m group we find: (x, y, 0) 42 − − − − − → (¯ y, x, 1/2) 42 − − − − − → (¯ x, ¯ y, 0) 42 − − − − − → (y, ¯ x, 1/2) m m m m (x, y, 0) 42 − − − − − → (¯ y, x, 1/2) 42 − − − − − → (¯ x, ¯ y, 0) 42 − − − − − → (y, ¯ x, 1/2), (45) and the (x, y, 0) has a multiplicity of only 4, half of the order of the factor group. c V. Lua˜ na, QTC Murcia 2008 (29)
list of special symmetry positions within a group (Wyckoff’s positions) is fully exploited for the description of crystal structures. In the case of P42/m group: Wyckoff Local symm. Equivalent positions 8k 1 (x, y, z), (¯ y, x, z + 1/2), (¯ x, ¯ y, z), (y, ¯ x, z + 1/2), (x, y, ¯ z), (¯ y, x, 1/2 − z), (¯ x, ¯ y, ¯ z), (y, ¯ x, 1/2 − z). 4j m.. (x, y, 0), (¯ x, ¯ y, 0), (¯ y, x, 1/2), (y, ¯ x, 1/2). 4i 2.. (0, 1/2, z), (1/2, 0, z + 1/2), (0, 1/2, ¯ z), (1/2, 0, 1/2 − z). 4h 2.. (1/2, 1/2, z), (1/2, 1/2, z + 1/2), (1/2, 1/2, ¯ z), (1/2, 1/2, 1/2 − z). 4g 2.. (0, 0, z), (0, 0, z + 1/2), (0, 0, ¯ z), (0, 0, 1/2 − z). 2f ¯ 4.. (1/2, 1/2, 1/4), (1/2, 1/2, 3/4). 2e ¯ 4.. (0, 0, 1/4), (0, 0, 3/4). 2d 2|m.. (0, 1/2, 1/2), (1/2, 0, 0). 2c 2|m.. (0, 1/2, 0), (1/2, 0, 1/2). 2b 2|m.. (1/2, 1/2, 0), (1/2, 1/2, 1/2). 2a 2|m.. (0, 0, 0), (0, 0, 1/2). Notice: when the coordinates of a transformed point are outside of the main cell we can put them back within 0 ≤ x, y, z < 1 by adding up integers. Example: x = 0.2 and y = 0.3 produce the next 4j positions: (0.2, 0.3, 0), (−0.2, −0.3, 0) = (0.8, 0.7, 0), (−0.3, 0.2, 0.5) = (0.7, 0.2, 0.5), and (0.3, −0.2, 0.5) = (0.3, 0.8, 0.5). c V. Lua˜ na, QTC Murcia 2008 (30)
The International Tables use a simple trick to reduce the list of equivalent positions on centered cells. Only the positions generated by the point group Gp are explicitely given, and the centering vectors must be added to them to produce the whole set of the factor group. P or R† I F A B C R‡ + (H) R‡ − (H) (0, 0, 0) (0, 0, 0) (0, 0, 0) (0, 0, 0) (0, 0, 0) (0, 0, 0) (0, 0, 0) (0, 0, 0) (1/2, 1/2, 1/2) (1/2, 1/2, 0) (0, 1/2, 1/2) (1/2, 0, 1/2) (1/2, 1/2, 0) (2/3, 1/3, 1/3) (2/3, 1/3, 2/3) (1/2, 0, 1/2) (1/3, 2/3, 2/3) (1/3, 2/3, 1/3) (0, 1/2, 1/2) R†: trigonal cell with rhombohedral axes. R‡(H): trigonal cell with hexagonal axes. Ex: In the case of the R32 group (Num. 155) the ITA gives: Wyckoff Sim. local Posiciones equivalentes 18f 1 (x, y, z), (¯ y, x − y, z), (y − x, ¯ x, z), (y, x, ¯ z), (x − y, ¯ y, ¯ z), (¯ x, y − x, ¯ z) 9e .2 (x, 0, 1/2), (0, x, 1/2), (¯ x, ¯ x, 1/2) 9d .2 (x, 0, 0), (0, x, 0), (¯ x, ¯ x, 0) 6c 3. (0, 0, z), (0, 0, ¯ z) 3b 32 (0, 0, 1/2) 3a 32 (0, 0, 0) and the three R(H) centering vectors must be added to complete the list. c V. Lua˜ na, QTC Murcia 2008 (32)
crystal structures Grouped by structure symbols. (other alternatives: http://cst-www.nrl.navy.mil/lattice/) A1 Cu, Cubic, a = 3.609 ˚ A, Fm¯ 3m, Z = 4. fcc Cu (4a) (0, 0, 0). A2 Li, Cubic, a = 3.46 ˚ A, Im¯ 3m, Z = 2. bcc Li (2a) (0, 0, 0). A3 Be, Hexag., a = 2.2860, c = 3.5843 ˚ A, (hcp ideal: c/a ≈ 1.63) P63/mmc, Z = 2. Be (2c) (1/3, 2/3, 1/4). A4 C (diamante), Cubic, a = 3.5667 ˚ A, Fd3m, Z = 8. C (8a) (1/8, 1/8, 1/8). A9 C (grafito), Hexag., a = 2.456, c = 6.696 ˚ A, P63/mmc, Z = 4. C (2b) (0, 0, 1/4); C (2c) (1/3, 2/3, 1/4). B1 NaCl, Cubic, a = 5.6402 ˚ A, Fm¯ 3m, Z = 4. Na (4a) (0, 0, 0); Cl (4b) (1/2, 1/2, 1/2). B2 CsCl, Cubic, a = 4.123 ˚ A, Fm¯ 3m, Z = 1. Cs (1a) (0, 0, 0); Cl (1b) (1/2, 1/2, 1/2). B3 β-ZnS (blenda), Cubic, a = 5.4060 ˚ A, F¯ 43m, Z = 4. Zn (4a) (0, 0, 0); S (4c) (1/4, 1/4, 1/4). B4 ZnO (zincita, wurtzita), Hexag., a = 3.2495, c = 5.2069 ˚ A, P63mc, Z = 1. Zn (2b) (1/3, 2/3, z ≈ 0); O (2b) (1/3, 2/3, z ≈ 0.345). c V. Lua˜ na, QTC Murcia 2008 (34)
A2: Li A3: Be A4: diamante A9: grafito B1: NaCl B2: CsCl B3: ZnS (blenda) B4: ZnO (wurtzita) Plots made with tessel (http://web.uniovi.es/qcg/tessel/tessel.html) and POVRay (http: //www.povray.org). c V. Lua˜ na, QTC Murcia 2008 (35)
Bases Cambridge Structural Database (CSD) (http://www.ccdc.cam.ac.uk/products/csd/): The main source of crystal structures for organic and organometallic compounds. Commercial, access by subscription. 436436 structures as of 2008-01-01. Inorganic Crystal Structure Database (ICSD) (http://www.fiz-karlsruhe.de/icsd.html?&L= 0): Natural and synthetic inorganic compounds. Commercial, access by subscription. > 100000 entries (feb 2007). See a fully functional subset at http://icsd.ill.fr/icsd/ (3592 samples). CRYSTMET (http://www.tothcanada.com/databases.htm): Metals, including alloys, inter- metallics and minerals. Commercial, access by subscription. 109877 entries (2006-11-10). American Mineralogist Crystal Structure Database (AMCSD) (http://rruff.geo.arizona. edu/AMS/amcsd.php): Minerals. Free access (financed by NSF). 3156 different minerals (many more entries, as a mineral may appear at different pressures and temperatures). Reciprocal Net (http://www.reciprocalnet.org/): Small but well choosen set of quite common molecules and materials. Data from CSD and other sources. Free access (financed by NSF). Protein Data Bank (PDB) (http://www.rcsb.org/pdb/): Structures of large biological molecules, including proteins and nucleic acids. Free access (international support). 48638 structures (2008-01-29). Indispensable portal for anyone working on biomolecules. c V. Lua˜ na, QTC Murcia 2008 (36)
of Proteins (SCOP) (http://scop.mrc-lmb.cam.ac.uk/scop/): Fur- ther analysis of the proteins contained in PDB: folds, superfamilies, evolutionary relationship, etc. Free access. Nucleic Acids Data Bank (NADB) (http://ndbserver.rutgers.edu/): Similar to PDB, but specialized on oligonucleotides. Free access (international support). 3745 structures (2008-01-17). Mineralogy Database (http://webmineral.com/): Good database on minerals and gems maintained by commercial dealers. Free access. Crystal Lattice Structures (http://cst-www.nrl.navy.mil/lattice/): Very good description of the common crystal lattice structures of the elements and simple compounds. Free access. Crystallography Open Database (COD) (http://www.crystallography.net/): Voluntary effort to provide an open alternative to CSD and ICSD. Absolutely free access. Some 48000 entries (dec 2006) and growing fast. A sister PCOD database specializes on theoretically predicted structures. Powder diffraction file (PDF) (http://www.icdd.com/): Largest DB on single phase powder diffraction pattern. Widely used to identify compounds based on their fingerprint spectra. Commercial. Database of Macromolecular Movements (http://molmovdb.mbb.yale.edu/molmovdb/): Anal- ysis and prediction of the dynamical behaviour of macromolecules. Movies, morphings, etc. Free access. c V. Lua˜ na, QTC Murcia 2008 (37)
any other good structures database? Please, e-mail me the address and details (mailto:[email protected]) References Ashcroft and Mermin, 1976 [1]; Bhadeshia, 2006 [2]; Bradley and Cracknell, 1972 [6]; Burns and Glazer, 1990 [3]; Giacovazzo et al., 2002 [4]; International Tables, vol A [5]. Exercises 1. The polymorph I of benzene has been measured again recently by A. Budzianowski and A. Katrusiak, Acta Cryst. B 62 (2006) 94. The observed structure is orthorhombic, space group Pbca (Num. 61), a = 7.287(6), b = 9.20(2), c = 6.688(9) ˚ A, four molecules per cell (Z = 4), and the atoms occupy the non-equivalent positions shown in the table. Using this data: (a) determine the crystal density; (b) obtain the average C-C and C-H distances, the average C-C-C, H-C-H, and H-C-C angles, and the average deviation from the planarity of the benzene units; (c) plot the benzene stacking using any molecular modeller. C −0,053 7 (8) +0,142 5 (9) +0,009 7 (12) H −0,085 (6) +0,246 (7) +0,034 (8) C +0,084 0 (7) +0,092 4 (10) +0,137 3 (10) H +0,140 (6) +0,156 (6) +0,219 (8) C −0,134 3 (7) +0,052 1 (9) −0,123 5 (12) H −0,220 (6) +0,080 (6) −0,204 (9) c V. Lua˜ na, QTC Murcia 2008 (38)
position of the Pbca group is: (1) (x, y, z), (2) (1/2−x, −y, 1/2+z), (3) (−x, 1/2+y, 1/2−z), (4) (1/2+x, 1/2−y, −z), (5) (−x, −y, −z), (6) (−1/2+x, y, −1/2−z), (7) (x, −1/2−y, −1/2+z), (8) (−1/2−x, −1/2+y, z). (A) Get the Seitz matrices of the group. (B) Obtain the multiplication matrix of the factor group and use it to determine the equivalence classes of the Seitz operators. Remember: two operations { ˆ R|ˆ tR} and { ˆ S|ˆ tS} are equivalent if there exists another operation { ˆ X|ˆ tX } in the group such that { ˆ X|ˆ tX }−1{ ˆ R|ˆ tR}{ ˆ X|ˆ tX } = { ˆ S|ˆ tS}. 3. Transform the F and I cells into their primitives. Obtain then the reciprocal cells of both primitives and use this to explain a typical sentence reproduced in most solid state textbooks: the reciprocal of a F cell is an I cell, and viceversa. 4. The non-orthogonal parallelepipedic cells are quite useful for most crystallographic tasks, but there are occasions in which a cartesian frame is far more convenient. Let us define one of the possible cartesian axes in the following way: First, the origin remains unchanged; the cartesian system is described by the three orthonormal vectors {i, j, k}; i is collinear with the crystallographic a vector; both, i and j lie in the ab plane; and, finally, i × j = k. Find the transformation from crystallographic to cartesian coordinates and viceversa, for a general crystal (a, b, c, α, β, γ). c V. Lua˜ na, QTC Murcia 2008 (39)
of Material Science The macroscopic behavior of matter (gases, liquids, solids, ...) is ruled by a thermodynamical potential. For an hydrostatic system under fixed pressure and temperature conditions, the ruling potential is the general Gibbs free energy: G (p, T; x) = E(V, T; x) + pV (x) − TS(V, T; x) = A(V, T; x) + pV (x), (54) where • p: pressure; V : volume; T: absolute (Kelvin) temperature; • x: variables describing the internal geometry of the system; • E: internal energy; S: entropy; A: Helmhotz free energy. Thermodynamic equilibrium conditions are found by minimizing G with respect to the internal variables: min x G (p, T; x) =⇒ G(p, T) = G (p, T; xmin ). (55) c V. Lua˜ na, QTC Murcia 2008 (41)
two parts: Equilibrium: (necessary cond.) The gradient of G with respect to the x coordinates must be zero: ∇xG (p, T; x) = 0 (56) The points fulfilling this equation are critical points of the G surface. • Stability: (sufficient cond.) Critical points can be classified according to the sign of the eigenvalues of the Hessian matrix: H = ∇ ⊗ ∇G =⇒ U−1 H U = Λ = diag(λ1, λ2, · · · ). (57) Minima: all the eigenvalues are positive; n-order saddle points: n of the eigenvalues are negative; maxima: all the eigenvalues are negative. The absolute minimum represents the most stable conformation. The other minima correspond to metastable conformations. First order saddle points are the transition states in a reaction pathway: the path being the gradient line going down from the saddle to two different minima. c V. Lua˜ na, QTC Murcia 2008 (42)
= A(V, T; x) + pV (x) A(V, T; x) = Emec(V ; x) + Atrasl(V, T; x) + Arot(V, T; x) + Avib(V, T; x) + · · · (58) • Quantum mechanics (QM): Emec is the eigenvalue of the system hamiltonian: ˆ HΨ = EΨ. Solving the time independent Schr¨ odinger equation is difficult and time consuming. • Molecular mechanics (MM): If quantum mechanics is too slow, we can resort to obtain Emec by adding up interaction potentials. • QM/MM: A combination of techniques (multiscale methods) is the basis to tackle very large systems. • Statistical mechanics: Provides the equation for the remaining terms. For a crystal under small harmonic vibrations, for instance: Avib ≈ ∞ 0 g(ν) hν/2 + RT ln(1 − e−hν/kBT ) dν. (59) The vibration frequencies and degeneration, ν and g(ν) respectively, are obtained from a quantum or molecular mechanics calculation. c V. Lua˜ na, QTC Murcia 2008 (43)
crystal phase The problem: Crystal space is too large to perform blind searches looking for stable structures. Strategy: Impose some symmetry on the cell parameters and the atomic occupancy. The restrictions define a structure subspace in which minimizations and derivatives are performed. Care must be taken to ensure that the resulting phases are truly stable (this may involve breaking the assumed symmetry). Example: ZnO, wurtzite phase, hexagonal, P63mc, Zn (2b) (1/3, 2/3, z ≈ 0), O (2b) (1/3, 2/3, zO) with zO ≈ 0,345. In this case x = (a, c, zO), and obtaining the equlibrium geometry requires a 3-parameter minimization. Pressure and temperature effects: It is simple to include pressure effects on the theoretical simulations but it is rather dificult to deal accurately with the temperature. Fortunately: (1) pressure can induce larger and more significant changes than temperature; and (2) experimentation on high pressure conditions is difficult and theoretical help is usually much welcomed. The static model: assume T = 0 K and ignore completely T effects, including zero-point contributions to Emec. Under this approximation: A(V, T; x) ≈ Ecell(V ; x), G (p, T; x) ≈ Ecell(V ; x) + pV (x). (60) Significant equilibrium properties: geometry (xeq); equation of state (EOS, p(V ), V (p), or f(p, V, T) = 0); bulk modulus (B0 = −V (∂p/∂V )T , and B(p)); elastic constants; ... c V. Lua˜ na, QTC Murcia 2008 (44)
elastic moduli). The elastic moduli are the curvatures of the elastic energy with respect to the strain components: cijkl = ∂2φ ∂ ij∂ kl , invariant under i ↔ j, k ↔ l, ij ↔ kl. (68) Strain components are adimensional. The magnitude of c’s is then energy/volume=pressure. The symmetry of the τ, and c tensors is made explicit by defining a new suffix notation: τ11 τ22 τ33 τ23 τ31 τ12 11 22 33 2 23 2 31 2 12 τ1 τ2 τ3 τ4 τ5 τ6 1 2 3 4 5 6 Strain non-diagonal terms are duplicated because they appear twice as many times than diagonal terms in the 65 equation. In this way, a cmn is exactly one of the cijkl and the φ = (1/2) i,j cij i j expression is maintained. Stress terms, however, are not duplicated, so the 2 factor is included into the s’s definition: s11 = s1111, but s16 = 2s1112, and s66 = 4s1212. The index change transforms and τ into six-element vectors, and the fourth rank tensors c and s into 6 × 6 symmetric matrices. In the worst case (a triclinic crystal), there are 21 independent elastic moduli. The unit cell symmetry can reduce much this number. Intrinsic stability of a phase. A crystal structure is said to be mecanically stable if ∇φ = 0 and the elastic energy is positive definite with respect to all strains, i.e. all the eigenvalues of the c matrix are positive. This would ensure the phase to be a true minimum on the energy surface at zero p and T. Were an eigenvalue negative or close to zero, its eigenvector would show how the c V. Lua˜ na, QTC Murcia 2008 (47)
to attain a lower energy minimum. The stability condition can be generalized to any p and T by using φ = G /V to compute the effective ceff ij . The stability condition impose restrictions on the elastic moduli. For instance, on a cubic crystal c11 > |c12| , c11 + 2c12 > 0, c44 > 0, (69) whereas for an hexagonal cell c11 > |c12| , (c11 + c12)c33 > 2c3 13 , c44 > 0, c66 > 0. (70) Elastic moduli and the crystal systems (See Bhagavantam [7] or Nye [8]) Cubic Hexagonal Orthorhombic c11 c12 c12 0 0 0 c11 c12 0 0 0 c11 0 0 0 c44 0 0 c44 0 c44 c11 c12 c13 0 0 0 c11 c13 0 0 0 c33 0 0 0 c44 0 0 c44 0 (c11−c12)/2 c11 c12 c13 0 0 0 c22 c23 0 0 0 c33 0 0 0 c44 0 0 c55 0 c66 For a isotropic medium c44 = (c11−c12)/2 (Cauchy relationship). c V. Lua˜ na, QTC Murcia 2008 (48)
using the crystal static deformation method pioneered by Catti [9, 10]. The only requirement is a computational method able to provide the crystal energy for arbitrary crystal geometries. Starting from an equilibrium configuration, the technique works by performing controlled deformations of the crystal. Each deformation is designed to produce a simplified form of the elastic energy 2φ = Tc . For instance, the deformation T = [η, 0, 0, 0, 0, 0] in a cubic crystal has an elastic energy 2φ = c11η2 and it is clear that c11 can be obtained as (∂2φ/∂η2). A short collection of calculations for some small values of η plus a least square fitting is enough to complete the task. This particular deformation transforms the unit cell from a cube to a square-based prism of dimensions [a(1+η), a, a, 90, 90, 90], thus lowering the symmetry from cubic to tetragonal. The symmetry reduction can have the added consequence that some of the atoms within the cell gain degrees of freedom and their position within the cell should be reoptimized for each new value of η. Failing to take into account this inner strain can produce a significant impact on the calculated value of the elastic moduli. Designing a set of sensible deformations can be a creative task with a large influence on the computational effort. The best routes will elude creating inner strain and lowering the unit cell symmetry as much as possible. The bulk modulus, B, is related to the elastic moduli. In a cubic system, for instance, B = (c11 + 2c12)/3. c V. Lua˜ na, QTC Murcia 2008 (50)
[fluorite; Fm¯ 3m (Num. 225); Ca (4a) (0, 0, 0); F (8c) (1/4, 1/4, 1/4)]. Strain Space G. Inner strain 2φ Cell [η, η, 0, 0, 0, 0] 4|mmm No 2(c11+c12)η2 [a(1+η), a(1+η), a, 90, 90, 90] [η, η, −2η, 0, 0, 0] 4|mmm No 6(c11−c12)η2 [a(1+η), a(1+η), a(1−2η), 90, 90, 90] [η, η, η, 0, 0, 0] Fm¯ 3m No 3(c11+2c12)η2 [a(1+η), a(1+η), a(1+η), 90, 90, 90] [0, 0, 0, η, η, η] R¯ 3m F (x, x, x) 3c44η2 [a, a, a, α, α, α], 2η = cos(90−α) x ≈ 1/4 Ca and F remain on a symmetry fixed position for the first three proposed deformations, and F has only one degree of freedom in the fourth one. [η, η, η, 0, 0, 0] is a breathing dilatation of the cubic unit cell and, consequently, the elastic energy is proportional to the bulk modulus. The c44 modulus could also be obtained through a [0, 0, 0, 0, 0, η] deformation, but this would reduce the cell to moclinic symmetry, instead of rhombohedral as the proposed [0, 0, 0, η, η, η]. The space group that results from each deformation is usually the highest subgroup of the original group cell that belongs to the final crystal system. The importance of the elastic characterization. Bulk modulus and elastic moduli are very important properties of the crystal beyond their significant role on establishing the stability of the crystal phase and helping to predict the possible deformation of the unit cell shape in a potential phase transition. Elastic data is required in crystallography to calculate the thermal diffuse scattering c V. Lua˜ na, QTC Murcia 2008 (51)
intensities. The interpretation of the observed seismic waves, that constitute our best probe on Earth innards, depend on the assumed elastic properties of the solid phases present in the earth mantle. Material scientist are engaged in a long quest for superhard materials that maintain their hardness on a high pressure, high temperature, or high radiation working ambient. References Bhagavantam, 1966 [7]; Nye, 1985 [8]; Catti, 1985 [9], 1989 [10]; Catti et al., 1991 [11]; Jona and Marcus, 2001 [12]; Ashcroft and Mermin, 1976 [1]. Exercises 1. Use the data below and the equations of state described previously to determine E0, V0, B0 and the appropriate number of derivatives of B for the bcc phase of lithium. Compare the values predicted by the different EOS models. Examine the stability of the Birch-Murnaghan results if the EOS is truncated to second and third order. Check the suitability of using five or six points centered around the minimum and a least squares fit to a parabola, as it is commonly done by some authors. Compare in a plot the original data and the diferent EOS. Caution and hints: Take care to convert appropriately the energy and lenght units. Report pres- sures and bulk moduli in GPa. The open source code gnuplot (http://www.gnuplot.info/) provides a robust an easy to use approach to perform non-linear least squares fitting for c V. Lua˜ na, QTC Murcia 2008 (52)
of MgO (aiPI calculations) Cubic, Fm¯ 3m, a = 4.210 ˚ A Mg 4a 0 0 0 O 4b 1/2 1/2 1/2 aiPI input: uchf crystal title MgO. Experimental geometry. spg f m -3 m cell 7.9557 7.9557 7.9557 90.0 90.0 90.0 neq 0.0 0.0 0.0 mg.ion mg.int mg.cint mg.lint neq 0.5 0.5 0.5 o2.ion o2.int o2.cint o2.lint endcrystal end The *.ion files contain a description of the basis set, electron occupancy, starting orbitals, etc, for each ion type in the crystal. CPU: ≈44 s per 100 geometries (Intel PIV/1.1 GHz). c V. Lua˜ na, QTC Murcia 2008 (55)
aiPI calculation provides Emec(V ). Under the static approximation (T = 0 K and the zero point energy is neglected) G ≈ Emec(V ) + pV and min a E → {ae, Ee}, Elatt = Ee − Evac(Mg+2) − Evac(O−1), (71) A = E + TS ≈ E, p = − ∂A ∂V T , Be = −V ∂p ∂V T ≈ V ∂2E ∂V 2 . (72) −720 −700 −680 −660 −640 −620 −600 −580 −560 3.5 4.0 4.5 5.0 5.5 Elatt (kcal/mol) a (Å) Prop. Unit aiPI Exptal. ae ˚ A 4.212 4.210 Elatt kcal/mol 708 725 Be GPa 156 163 (dB/dp)e — 3.28 4.13 Cp J/mol K 38.16 37.89 α 10−6 K−1 24.8 13.5 Thermal properties (Cp and α = V −1(∂V/∂T)p) have been obtained using a quasiharmonic Debye model[14], in which the volume dependent bulk modulus, B(V ), is used to determine Debye’s temperature, θD = hνD/kB, as a function of volume. c V. Lua˜ na, QTC Murcia 2008 (56)
Some results on LiCl: 5.0 5.5 6.0 6.5 7.0 7.5 8.0 50 60 70 80 90 100 -0.32 -0.30 -0.28 -0.26 -0.24 -0.22 E (hartree) a (bohr) α (deg) E (hartree) The quantum-mechanical energy, E(a, α), is the po- tential energy surface that determines stability under 0 K. The effect of pressure is obtained by adding the work term , pV . Temperature effects have been esti- mated using a simple quasiharmonic Debye model. B1 and B2 phases are metastable as far as G (x; p, T) is a minimum at their corresponding positions in the phase space. Of particular importance is the curvature represented by the ceff 44 elastic constant. Remember: ceff ij = 1 V ∂G ∂εi∂εj (73) where εi are the Voigt-Lagrange deformation parame- ters. In particular ε4 = 2ε23, and c44 is the curvature with respect to the Buerger’s R¯ 3m opening. -100 0 100 200 300 400 500 600 700 0 50 100 150 200 250 300 c44, c44 eff (GPa) P (GPa) B1 B1(eff) B2 B2(eff) c V. Lua˜ na, QTC Murcia 2008 (58)
A reaction path, (a, α) within Buerger’s mechanism, has been obtained by minimizing G (x; p, T) for fixed values of α. Pathways obtained for different pressures are almost parallel. This is a consequence of the sym- metry of the hessian matrix, H = ∇ ⊗ ∇G , at both metastable phases. 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 50 60 70 80 90 100 a (bohr) α (grados) 0 GPa 40 GPa 80 GPa 300 GPa c V. Lua˜ na, QTC Murcia 2008 (59)
-0.06 -0.04 -0.02 0.00 0.02 0.04 0.06 0.08 0.10 50 60 70 80 90 100 110 G (hartrees) α (grados) 0.00 GPa 40.00 GPa 80.00 GPa 300.00 GPa The free energy profile along the reaction pathway, G(α), determines the most stable phase and the ki- netical aspects of the phase transition. The B1-B2 transition pressure is Ptr ≈ 80 GPa in the equilibrium (LiCl), but the transition barrier is large and different for the compression-decompression steps: significant hysteresis should be expected. c V. Lua˜ na, QTC Murcia 2008 (60)
0 500 1000 1500 2000 2500 0 50 100 150 200 T(K); ∆G‡ (K) P (GPa) B1 → B2 B2 → B1 A qualitative model for the Hysteresis can be expressed in simple kinetical terms: dnB1 dP = ωB1e−∆G‡ B1→B2 /kBT nB1 − ωB2e−∆G‡ B2→B1 /kBT nB2, (74) where nB1 and nB2 are the concentrations of both phases, ∆G‡ are the energy barriers (different for the B1 → B2 and B2 → B1 processes except at equilib- rium), and the ωi are unknown kinetical constants. B1-B2 equilibrium pressures at 300 K. Ptr F Cl Br I Li 252 79 94 113 Na 12 (23–24) 21 (26–30) 16 16 K 5.6 (1.73) 2.0 (1.9–2.1) 1.64 (1.77) 2.7 (1.8–1.9) Rb −0.2 (0.9–3.3) 0.3 (0.5) 0.1 (0.45) 0.8 (0.3–0.4) Cs −2.6 (2) −1.3 (B2) −1.5 (B2) −0.7 (B2) c V. Lua˜ na, QTC Murcia 2008 (61)
[16]; Lua˜ na and Pueyo, 1990 [17]; Lua˜ na and Fl´ orez, 1992 [18]; Lua˜ na, Fl´ orez, and Pueyo, 1993 [19]; Pend´ as et al., 1994 [20]; Blanco, 1997 [21]; Schlick, 2002 [22]. Exercises 1. The contiguous figure represents the energy of the CaO crystal at the B1 and B2 phases calculated by Mota et al. [23]. Determine the equilibrium transition pressure. 2. Search the recent literature (e.g. 2000 to current date) on the B1-B2 phase transition and write a short resume of the most relevant experimental and theoretical advances on the subject. c V. Lua˜ na, QTC Murcia 2008 (62)
Let assume the system to be made of N interacting bodies (neutral atoms or charged ions, usually: atoms, from now on, to simplify). The mechanical energy can be obtained as: Emec = N i=1 v1(ri) one body + N i>j v2(ri, rj) two body + N i>j>k v3(ri, rj, rk) three body +... (75) where i>j sums over all different pairs of bodies, i>j>k over all different triplets, and so on. The properties of ordinary matter are controlled by the kinetic energy and the electrostatic interaction of the fundamental particles (electrons and nuclei). The two-body nature of the Coulomb law makes only natural that the v2(ri, rj) ≈ v2(rij) terms dominate the mechanical energy. The required self-consistency between the quantum mechanical wavefunction and the potencial acting on the particles can be viewed as the origin of the (N = 2)-body terms. In other words, the local environment of a body is affected by its neighbors and this modifies the way in which they interact. In any way, the many-body terms are responsible for many of the fine details that determine phase stability and thermodynamical properties. c V. Lua˜ na, QTC Murcia 2008 (64)
of atoms Dealing with the energy of atoms and groups of atoms will be simpler by defining the next three quantities: • The effective energy of an atom i includes all the contributions to the total energy of the whole system in which the atom i participates: Ei eff = v1(ri) V i 1 + N j(=i) v2(ri, rj) V i 2 + N j>k(=i) v3(ri, rj, rk) V i 3 + . . . (76) where V i n describes the sum of all the n-body terms that contain i. As a consequence of this definition, the minimization of Ei eff with respect to the position and internal structure of i is equivalent to the minimization, respect to the same variables, of the total energy of the system, Emec [PRB 39 (1989) 11093]. This idea comes from McWeeny’s and Huzinaga’s Quantum Electronic Separability Theory, where it has been formulated as a Restricted Variational Principle. c V. Lua˜ na, QTC Murcia 2008 (65)
of i contains an even part of all the energy terms in which the atom is included. Accordingly, the V i n n-body term is multiplied by an 1/n factor: Ei add = V i 1 + 1 2 V i 2 + 1 3 V i 3 + · · · + 1 n V i n + . . . (77) The additive energies can be added together to give the total energy of the system: Emec = i Ei add . (78) The additive energy of an arbitrary subset of atoms can be also formed easily: Eadd(AxByCz . . . ) = xEA add + yEB add + zEC add + . . . (79) • The local energy of i within G is a quantity created to help in the calculation of the effective energy of a group of atoms. We want to do EG eff = i∈G Ei(G) loc . (80) Therefore, Ei(G) loc has to be additive within G but effective with respect to the rest of the system. The interaction of i with jkl . . . must be shared evenly only between the atoms that c V. Lua˜ na, QTC Murcia 2008 (66)
97 (1992) 6544]. In other words: Ei(G) loc = v1(ri) + j(=i) 1 nG ij v2(ri, rj) V i(G) 2 + j>k(=i) 1 nG ijk v3(ri, rj, rk) V i(G) 3 + . . . (81) where nG ijk... is the number of atoms among i, j, k, ... that belong in G. The calculation of Ei(G) loc is rather cumbersome, as it depends on both, the atom i and the group G. When G is small it may be more convenient to get the V i(G) n terms as the difference between the general V i n minus a local correction. For the two and three-body terms we can use: V i(G) 2 = V i 2 − 1 2 (=i) j∈G v2(ri, rj), (82) V i(G) 3 = V i 3 − 2 3 (=i) (j>k)∈G v3(ri, rj, rk) − 1 2 (=i) j∈G j / ∈G v3(ri, rj, rk). (83) The formulas of four and further body terms become progressively more complicated. c V. Lua˜ na, QTC Murcia 2008 (67)
useful on the treat- ment of periodic structures. The cell energy of a crys- tal, for instance, is just the additive energy of the cell group: Ecell = Ecrystal ncell ≡ Ecell add = i∈cell Ei add . (84) Notice that for an infinitely periodic model of a perfect crystal Ecrystal and ncell are both inifinite but Ecell is a finite and well defined quantity. The effective energy of a group, on the other hand, is the quantity to determine on embedded cluster model calcula- tions, i.e. on the calculation and optimization of a cluster of atoms embedded into a lattice that is assumed to remain frozen: Ecluster ≡ Ecluster eff = i∈cluster Ei(cluster) loc . (85) Cluster Frozen lattice c V. Lua˜ na, QTC Murcia 2008 (68)
potential: When the atoms carry net charges, the electrostatic charge-charge interaction provides a leading contribution to the energy of the system. The term ionic matter alludes to the leading role of this term. v2(rij) = qiqj 4πε0rij . (86) The summation of the Coulomb terms on an infinite system, like a crystal, gives rise to a conditionally convergent term that needs special techniques. Lennard-Jones potential: [John Edward Lennard-Jones, Trans. Faraday Soc. 25 (1929) 668] −1.0 −0.9 −0.8 −0.7 −0.6 −0.5 −0.4 −0.3 −0.2 −0.1 0.0 1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4 U(r) / ε r / σ De A −r−6 term describes the long-range London dispersion attraction. The +r−12 term emulates the short-range re- pulsion between atoms. The LJ6-12 term is widely used in modelling the van der Waals interactions: U(r) = 4 σ r 12 − σ r 6 , ( , σ ∈ R > 0). (87) The equation can be transformed into a universal form by using y = U/ and x = r/σ. Equilibrium properties: re = σ 6 √ 2, Ue = − , U∞ = 0, ke = 57,146 437 . . . /σ2. c V. Lua˜ na, QTC Murcia 2008 (69)
Morse, Phys. Rev. 34 (1929) 57] 0.00 0.20 0.40 0.60 0.80 1.00 0.6 0.8 1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4 U(r) / ε r / σ ρ=3 ρ=6 ρ=9 Proposed as a reasonable fit of the nuclear potential curves of diatomic molecules: U(r) = 1 − eρ(1−r/σ) 2 , ( , ρ, σ ∈ R > 0). (88) Conversion to a universal form: y = U/ and x = r/σ, with ρ left as a free parameter. Equilibrium properties: re = σ, Ue = 0, U∞ = , De = , ke = 2 ρ2/σ2. Common transformations: U(r) − , (ρ/σ) = α. Generalized Morse potential: Several generalizations have been proposed. For instance: U(r) = eqρ(1−r/σ) − qeρ(1−r/σ) , (q > 1). (89) Conversion to a universal form: y = U/ and x = r/σ, with ρ and q left as free parameters: ρ influences the curvature at the minimum and q the well depth. Equilibrium properties: re = σ, Ue = (1−q), U∞ = 0, De = (q−1), ke = q(q−1) ρ2/σ2. −1.60 −1.40 −1.20 −1.00 −0.80 −0.60 −0.40 −0.20 0.00 0.6 0.8 1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4 U(r) / ε r / σ ρ=6, q=1.5 ρ=6, q=2.0 ρ=6, q=2.5 c V. Lua˜ na, QTC Murcia 2008 (70)
an improvement of LJ6-12 potential, it con- serves the −r−6 London attraction, but changes the +r−12 respulsive wall into an exponential term, thus the frequent exp-6 nickname: U(r) = α α−6 6 α eα(1−r/σ) − σ r 6 for r>rmin, ∞ for r≤rmin. (90) −1.0 −0.5 0.0 0.5 1.0 0.5 1.0 1.5 2.0 2.5 (2/π) atan[U(r) (α−6)/ αε] r / σ rmin re Buckingham: α=9.3 U(r) U’(r) 0.0 0.2 0.4 0.6 0.8 1.0 6 8 10 12 14 rmin / σ α The internal cutoff rmin is defined as the smallest positive value for which U (r) = 0 (a maximum of the potential) and it helps to prevent the U → −∞ collapse at very short distances. Transformation into a universal form: y = U(α−6)/α and x = r/σ. The free parameter must be α > 7 or the potential would have no minimum. Equilibrium properties: re = σ, Ue = − , U∞ = 0, De = , ke = α(α−7) /(α−6)σ2. Internal cutoff: The iterative equation x = [eα(x−1)]1/7 converges to xmin starting from x ≈ 0.9. Then rmin = xminσ. Generalization: Sometimes the London term is seen as −C(σ/r)6. c V. Lua˜ na, QTC Murcia 2008 (71)
homogeneous parameters can be used to estimate the potential parameters for the heterogeneous combinations. Distances are combined as arithmetic means: σij = (σii +σjj)/2. Energies and energy-like parameters are combined as geometric means: ij = √ ii jj, αij = √ αiiαjj. Cutoffs and switching functions Most simulations define a cutoff radius beyond which the atom-atom interactions are assumed to be null. To avoid discontinuities in the energy, that can bring havoc to the simulation, the cutoff can be introduced by means of a switching function that ensures the analyticity of the product U(r)S(r). A standard cubic switch is: S(r) = 1 if r ≤ rsw, (rcut+2r−3rsw)(rcut−r)2 (rcut − rsw)3 if rsw<r≤rcut, 0 if r > rcut, (91) where rcut is the cutoff radius and rsw ≈ 0.95rcut is the switching radius. 0.0 0.2 0.4 0.6 0.8 1.0 2.80 2.85 2.90 2.95 3.00 3.05 3.10 S(r) r / σ rcut rsw c V. Lua˜ na, QTC Murcia 2008 (72)
There are many ways to introduce these terms. Bond angle potentials, for instance, can introduce a penalty when an angle θijk, between bonded atoms, deviates from some expected value: v3(i, j, k) = Kh(θijk − ¯ θj)2, or v3(i, j, k) = Kt(cos θijk − cos ¯ θj)2, (92) where the expected value ¯ θj depends on the assumed properties of the middle atom j. The bond angle arrangement of an atom is assumed to be governed by its Lewis pairs. For instance, we should expect 180 ◦ around a sp-C atom, 120 ◦ around a sp2-C, 109,47 ◦ around a sp3-C, and so on. This scheme can be augmented to include penalties for the deviation of a torsional angle from some expected value. A different strategy is including potentials between non-bonded next nearest neighbors (nnn), nnnn, and so for. The development of such a molecular mechanics force field is a quite specialized tasks that involves, first, developing the force field constants and, second, assesing its suitability for a given collection of compounds. c V. Lua˜ na, QTC Murcia 2008 (73)
is a quite successful approximation introduced for the dynamical treatment of pure and defective ionic crystals. Going beyond the point charge approximation by introducing dipole terms produces an intrinsic instability (polarization catastrophe) unless the polarizability decreases with the increased ionic overlap. This is automatically accounted for in the shell model by using two different entities to represent every atom/ion: a positively charged core that contains all atom mass, and negatively charged massless shell. Core and shell from the same atom interact through a strong harmonic potential. Shell-shell interactions are described by an appropriate two body potential. Core-core inter- actions are usualy neglected. kx2 Vij +q m −q Julian Gale’s Gulp code [16] (www.ivec.org/GULP) implements a huge collection of potential schemes, including the shell model, and a variety of dynamical methods to determine the properties of pure and defective crystals. GULP is the last of a large family of codes developed under the influence of the english Atomic Energy Authority at Harwell. The code is available upon request for academic usage. c V. Lua˜ na, QTC Murcia 2008 (74)
[16]; Lua˜ na and Pueyo, 1990 [17]; Lua˜ na and Fl´ orez, 1992 [18]; Lua˜ na, Fl´ orez, and Pueyo, 1993 [19]; Schlick, 2002 [22]. Exercises 1. The typical LJ6-12 potential is sometimes assumed to have a too strong short range repulsion and it is therefore changed into LJ6-11 or LJ6-10. Compare the behaviour of the three potentials. Determine, in general, the equilibrium properties of the LJn-m potential if n < m. 2. Do a quantum mechanical calculation of the binding energy of the Ne2 molecule. Check the influence of the correlation energy and the basis set. Fit the LJ and Morse potentials to your best results. 3. The “Embedded Atom Method” (EAM) [24, 25] is a popular and successful technique in developing interaction potentials for metallic systems. Write a short resume on the foundation of the method and some recent applications of the technique. c V. Lua˜ na, QTC Murcia 2008 (76)
The Lennard-Jones LJ6-12 internuclear potential Proposed in 1929 by John Edward Lennard-Jones [Trans. Faraday Soc. 25 (1929) 668]: U(r) = 4 σ r 12 − σ r 6 y=U/ =⇒ x=r/σ y(x) = 4(x−12 − x−6) (93) The scaling of U and r transforms the potential into a universal function y(x). LJ6-12 has been a favorite for the simulation of gases and liquids. The potential works well on closed shell atoms, particularly noble gases. −1.0 −0.9 −0.8 −0.7 −0.6 −0.5 −0.4 −0.3 −0.2 −0.1 0.0 1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4 U(r) / ε r / σ De /kB (K) σ (˚ A) He 10,2 2,28 Ne 47,0 2,72 Ar 119,8 3,41 Kr 164,0 3,83 H 8,6 2,81 C 51,2 3,35 N 37,3 3,31 O 61,6 2,95 F 52,8 2,83 (Allen & Tildesley, 1989) c V. Lua˜ na, QTC Murcia 2008 (78)
Simulation of the face centered cubic (fcc) structure The fcc structure will serve to illustrate the calculation of a crystal phase: Cell: (a, a, a, 90◦, 90◦, 90◦). Space group: cubic, Fm¯ 3m (No. 225). Atoms in the main cell: Z = 4, 1 (0, 0, 0), 2 (1/2, 1/2, 0), 3 (1/2, 0, 1/2), 4 (0, 1/2, 1/2). Metric matrix: G = a2 0 0 0 a2 0 0 0 a2 . Cell volume: V = G = a3. Interatomic distance: r2 ij = rT ij G rij = a2(x2 ij + y2 ij + z2 ij ) where rij = rj − ri. Scaling: Length: r = r/σ; Energy: E = E/ ; Pressure: p = pσ3/ ; . . . Cutoff and switch function: The r−6 term dominates the long range tail of the potential. To cut contributions < 10−n the cutoff radius must be rcut = 10n/6σ. No switching off will be used. Cell energy: Ecell= Z i=1 Ei add , where Ei add = 1 2 j(=i) ULJ(rij) and j runs over all atoms with rij ≤ rcut. c V. Lua˜ na, QTC Murcia 2008 (80)
Strategy: How to run over shells of neighbor cells in order of increasing distance to the main cell? 0 1 2 3 −2 −3 −1 0 +1 +2 +3 −3 −2 +1 +2 +3 −1 0 j i Caution: Many quite small contributions are added to larger values, producing large truncation errors. How to avoid the problem? Ecell = 0 do iat = 1, nat Eadd(iat) = 0; Econtri=0; L=0; doagain=.TRUE. while (doagain .and. L.le.100) do i = -L, L do j = -L, L if (abs(i).eq.L .or. abs(j).eq.L) then do jat = 1, nat xij = x(jat) + i - x(iat) yij = y(jat) + i - y(iat) rij = distance(xij, yij) if (rij.gt.0d0 .and. rij.le.rcut) then Econtri = Econtri + 0.5d0 * ULJ(rij) endif enddo endif enddo enddo Eadd(iat) = Eadd(iat)+Econtri L = L+1; doagain = (abs(Econtri) .ge. 1d-6) enddo Ecell = Ecell + Eadd(iat) enddo c V. Lua˜ na, QTC Murcia 2008 (81)
Strategy: Many tasks involve obtaining the energy for a collection of geometries. Much effort can be saved by keeping intermediate quantities between successive calculations. Ecell = Z i=1 4 2 j(=i) σ ahij 12 − σ ahij 6 = σ a 12 2 Z i=1 j(=i) h−12 ij A12 − σ a 6 2 Z i=1 j(=i) h−6 ij A6 , (99) where hij = (x2 ij + y2 ij + z2 ij ). The index i runs over the atoms in the main cell, but j sums all atoms in the whole crystal until convergence is achieved. However, once A12 and A6 are obtained, the optimization of a is a very simple analytical calculation: Y = A12X−12 − A6X−6 Y = 6X−7[−2A12X−6 + A6] Y = 156A12X−14 − 42A6X−8 Minimun: Y =0 ⇒ Xmin = amin σ = 2A12(α) A6(α) 1/6 , (100) where Y = Ecell/ and X = a/σ. Then: Ymin = −A2 6 /4A12, Ymin = 156A12X−14 min − 42A6X−8 min . In the case of a Lennard-Jones fcc crystal: A6 = 925,050 946 665 605 and A12 = 6 211,522 660 630 82. The equilibrium properties are then: amin = 1,541 737 σ, Vmin/Z = 0,916 159 770 σ3, Emin/Z = −8,610 200 154 , and Bmin = −V (∂p/∂V ) = 75,185 141 /σ3. c V. Lua˜ na, QTC Murcia 2008 (82)
Pressure effects: There are two basic ways of determining the effects of hydrostatic pressure (i.e. pressure applied isotropically) on the crystal properties: Method 1: the equilibrium pressure, as a function of the volume, can be determined as the slope of the A(V, T) function: p(V ) = − ∂A ∂V T . (101) Method 2: the equilibrium volume, as a function of the pressure, can be determined by minimizing the general Gibbs free energy: min V G (p, T; V ) = G(p, T) ⇒ V (p), (102) where G(p, T) = A(V, T) + pV . Both ways provide the static Equation of State (EOS), V (p) or p(V ), and the crystalBulk Modulus: BT (p) = −V ∂p ∂V T = V ∂2A ∂V 2 T . (103) The static approximation consists on the complete neglect of thermal effects (i.e. T = 0 K), even of the zero point vibrational energy. Using this approximation: A(V, T) ≈ Ecell(V ), G(p, T) ≈ Ecell(V ) + pV. (104) c V. Lua˜ na, QTC Murcia 2008 (83)
The pressure equation, 101, can be evaluated analytically for the Lennard-Jones fcc crystal: p ≈ − ∂Ecell ∂V = − Y ∂(Ecell/ ) ∂X σ−1 ∂X ∂a (3a2)−1 ∂V ∂a −1 = − σ3a2 Y = σ3 (4A12X−15−2A6X−9). (105) This equation provides the pressure as a function of the reduced length, X = a/σ. It is easy to transform it into a p(V ) relationship: p = p σ−3 = 4A12(V )−5 − 2A6(V )−3 (106) where V = V/σ3 is the reduced volume. A similar treatment can be applied to eq. 103. This gives rise to the next form of the reduced bulk modulus B = B σ−3 = 52A12 3(V )5 − 16A6 3(V )3 . (107) Both relationships, p (V ) and B (V ), can be used together to produce an implicit description of the more interesting B versus p relationship. c V. Lua˜ na, QTC Murcia 2008 (84)
0 20 40 60 80 100 0.6 0.7 0.8 0.9 1.0 1.1 1.2 1.3 1.4 p / (εσ−3) Vcell / (Zσ3) V0 / (Zσ3) 0.70 0.75 0.80 0.85 0.90 0.95 1.00 0 20 40 60 80 100 V / V0 p / (εσ−3) Pressure contraction: The cell volume dimin- ishes as the applied pressure increases. The ef- fect is highly non-linear, being dominated, for small volumes, by the p ∝ (V )−5 term. A small negative pressure could produce large cell expansions. A pressure of 100 /σ3 contracts the cell to some 70 % of its V0 = V (p=0) value. The cell energy versus volume follows a curve similar to the Lennard-Jones potential. Notice, however, that Ecell is a summation of many in- teraction energies. The cohesive energy, i.e. the energy needed to separate the crystal into non-interacting atoms, is −8,610 200 154 per atom. -8 -6 -4 -2 0 2 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 Ecell / (Zε) Vcell / (Zσ3) V0 /(Zσ3) Ecoh /(Zε) c V. Lua˜ na, QTC Murcia 2008 (85)
0 10 20 30 40 50 0.5 1 1.5 2 2.5 H* / Z V* / Z p* = 0 5 10 15 20 The enthalpy is the static approximation to the Gibbs free energy. The minimum of each H (V ) curve deter- mines the equilibrium properties for that par- ticular pressure. Whereas the p=0 curve has a null slope asymp- tote, the asymptotes for the other curves cor- responds to the pV ≡p V term. Bulk modulus increases with pressure, showing that compressing the crystal becomes progres- sively harder as the crystal shrinks. Bulk modulus is loosely related to the crystal hardness. The B(p) relationship is almost linear: B(p)/ σ−3 = 75,37 + 7,160p − 0,056 1p2 + ... 70 80 90 100 110 120 130 140 0 2 4 6 8 10 B / (εσ-3) p / (εσ-3) c V. Lua˜ na, QTC Murcia 2008 (86)
crystal structures Generalization for arbitrary crystal structures The equations developed for the fcc crystal can be generalized to any crystal structure. Cell energy: E = Ecell = A12 X12 − A6 X6 , X= a σ , A12 = 2 Z i=1 j(=i) h−12 ij , A6 = 2 Z i=1 j(=i) h−6 ij . (108) Cubic cells: hij = [x2 ij + y2 ij + z2 ij ]1/2. Tetragonal cells: hij = [x2 ij + y2 ij + r2 c z2 ij ]1/2, with rc = c/a. Hexagonal cells: hij = [x2 ij + y2 ij − xijyij + r2 c z2 ij ]1/2, with rc = c/a. Rhombohedric cells: hij = [(x2 ij + y2 ij + z2 ij ) + 2 cos α (xijyij + yijzij + zijxij)]1/2. Orthorhombic cells: hij = [x2 ij + r2 b y2 ij + r2 c z2 ij ]1/2, with rb = b/a, and rc = c/a. Monoclinic cells (β = 90◦): hij = [x2 ij + r2 b y2 ij + r2 c z2 ij + 2rcxijzij cos β]1/2. Triclinic cells: hij = [x2 ij +r2 b y2 ij +r2 c z2 ij +2rbxijyij cos γ+2rbrcyijzij cos α+2rczijxij cos β]1/2. c V. Lua˜ na, QTC Murcia 2008 (87)
crystal structures Equilibrium volume: The p (V ) relationship is highly non-linear and it cannot be directly inverted. However, the following iterative equation can be used to produce a series of values, {V0 ≈1, V1 , V2 , ...}, that converges to the equilibrium volume for a given pressure: Vi+1 = 4A12A4 3 p + 2A6A2 3 (Vi )−3 1/5 . (111) General Gibbs free energy: G ≈ H (V ; p ) = H = A12X−12 − A6X−6 + p A3X3 = A12 A3 V 4 − A6 A3 V 2 + p V . (112) It must be noticed that this is not the equilibrium value, that would be a funcion of just the pressure and temperature. The equilibrium value for G is easily obtained by using the equilibrium volume or the equilibrium pressure on the above equation. For instance: Heq (V ) = 5A12 A3 V 4 − 3A6 A3 V 2 . (113) c V. Lua˜ na, QTC Murcia 2008 (89)
crystal structures -10 0 10 20 30 40 50 60 70 0 5 10 15 20 25 30 35 40 45 50 H* / Z p* fcc bcc sc ideal hcp diamond -9 -8 -7 -6 -5 -4 0 1 2 3 4 5 Fcc and the ideal hcp structure have the same equilibrium enthalphy under any pressure. They are the most stable phases under any p >0 pressure. The sequence of stability follows an order that do not changes with pres- sure: Hhcp ≈ Hfcc < Hbcc < Hsc < Hdiamond . c V. Lua˜ na, QTC Murcia 2008 (92)
crystal structures 0 20 40 60 80 100 120 140 0 2 4 6 8 10 B* p* fcc bcc sc ideal hcp diamond Fcc and the ideal hcp structure have also the same bulk modulus under any hydrostatic pressure. The bulk modulus of every structure increases almost linearly with pres- sure. The sequence of hardness do not changes with pressure: Bhcp ≈ Bfcc > Bbcc > Bsc > Bdiamond. c V. Lua˜ na, QTC Murcia 2008 (93)
crystal structures 24 48 72 sc 0 1 2 3 4 5 24 48 72 bcc 24 48 72 fcc Radial neighbor density: g(r) 24 48 72 ideal hcp 0 24 48 72 diamond 0 1 2 3 4 5 r / σ The nearest neighbors (nn) shell appears at almost the same dis- tance for all the struc- tures. Fcc and hcp appear identical for the first two shells (nn and nnn), but start to dif- fer on the third shell (nnnn) of neighbors. c V. Lua˜ na, QTC Murcia 2008 (94)
crystal structures 4 8 16 32 64 128 256 512 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 G(r) r / σ fcc bcc sc ideal hcp diamond The accumulated number of neighbors, G(R) = R 0 g(r)dr (notice the log2 G scale), shows the close relationship be- tween the fcc, bcc and ideal hcp struc- tures. Fcc and hcp are identical on the nn and nnn shells. Fcc, hcp and bcc tend to be equivalent, on average, at large distances. The sc and diamond structures are far less dense. The stability of the crystal structures follows the order of the nn coordination index: 12(fcc and hcp) > 8(bcc) > 6(sc) > 6(diamond). The Lennard-Jones solid shows a clear preference for the dense structures. Is this a necessary consequence of the radial dependence of the two-body LJ potential? c V. Lua˜ na, QTC Murcia 2008 (95)
hcp crystal Treatment of the hcp crystal The geometry of all the crystal structures examined up to now depended on a single internal variable, a, that we converted into X after scaling. Determining the equilibrium geometry and properties involved minimizing the general Gibbs energy, G (p, T, X), with respect to this single variable. For most crystal structures, however, the geometry depends on a number of cell parameters and atomic cell positions, and the minimization of G (p, T, x) with respect to all of them requires some techniques beyond those used up to now. Let us start considering the general hcp structure: Cell: (a, a, c, 90, 90, 120). Metric matrix: G = a2 −a2/2 0 −a2/2 a2 0 0 0 c2 . Cell volume: V = √ 3 2 a3rc. Space group: P63/mmc (194). Atoms in the main cell: Z = 2; (2c) : (1/3, 2/3, 1/4), (2/3, 1/3, 3/4). Interatomic distance: r2 ij = rT ij G rij = a2 x2 ij + y2 ij − xijyij + r2 c z2 ij where rc = c/a and rij = rj − ri. We have already examined this structure, but only when the rc = 8/3 ratio was enforced. We will examine now the dependence of the energy and crystal properties on the two geometrical parameters: a and c or rc. c V. Lua˜ na, QTC Murcia 2008 (96)
the crystal From clusters to the crystal −9 −8 −7 −6 −5 −4 −3 −2 −1 0 0 100 200 300 400 500 600 700 800 900 1000 Eopt /n (ε) n (LJ atoms) The properties of LJ clusters should converge to the crystal properties as the cluster size increases. The convergence is, however, quite slow. Nanomatter prop- erties significantly differ from bulk range properties. c V. Lua˜ na, QTC Murcia 2008 (98)
the crystal The significant contribution of the surface and the accusated tendency to form an icosahedral arrangement are the main cause for the slow convergence of the clusters towards the bulk. LJ135 LJ147 LJ549 LJ557 LJ900 LJ906 LJ907 LJ910 c V. Lua˜ na, QTC Murcia 2008 (99)
et al. [26]; Doye & Calvo, 2002 [27]; Noya & Doye, 2006 [28]; Stillinger, 2001 [29]; Somasi et al., 2000 [30]. Exercises 1. Analyze the energy of the LJ crystal in the graphite phase as a function of the a and c lattice parameters. Determine the best rc = c/a ratio as a function of the external pressure. Compare the relative stability and hardness of the graphite and diamond phases. Determine the neighbor shell structure for graphite and compare it with the phases represented in the figure of page 94. 2. Determine the value of the independent elastic moduli for a Lennard-Jones solid in the sc, bcc, fcc, hcp, graphite and diamond phases. Compare the prediction of the model with the experimental values for noble gases, alkaline and alkaline-earth metals, the diamond and graphite phases of C and Si, etc. Cite the sources of your data and discuss the successes and failures of the LJ model. 3. Check very carefully the relative stability of the fcc and ideal hcp compact phases. Which is most stable? Is this result modified if the (σ/r)12 repulsion term is changed into (σ/r)m with m = 8, 9, 10, · · · c V. Lua˜ na, QTC Murcia 2008 (100)
fcc and the ideal hcp are just two of the infinite compact structures formed by stacking layers of close contact hard spheres. A detailed description of the close-packed structures can be found in Krishna & Pandey [31] (http://www.iucr.org/iucr-top/comm/cteach/ pamphlets/5/5.pdf). Check the relative stability of A, AB (hcp), ABC (fcc), ABA, ABAC, etc LJ6-12 crystal. 5. Use a Morse potential to examine the relative stability of the compact phase as a function of the internal ρ parameter. c V. Lua˜ na, QTC Murcia 2008 (101)
We want to solve the Hartree-Fock or Kohn-Sham equation for an electron in the average field of all other (infinite) crystal charges: − 2 2me ∇2 r + G(r, [ρ]) ψλ (r) = λ ψλ (r) (115) where G(r, [ρ]) = ˆ Vn + ˆ VH + ˆ Vxc : • ˆ Vn : electrostatic potential of the nuclei (or sum of the pseudopotentials of the chemical kernels); • ˆ VH (r) = R3 ρ(r )dr |r − r | : Coulomb potential due to the (posibly valence) electron density. Hartree term. • ˆ Vxc : Exchange and correlation term. Common approaches: Hartree-Fock (HF), LDA or LSDA (Local [Spin] Density Approach), GGA (Generalized Gradient Approach), . . . c V. Lua˜ na, QTC Murcia 2008 (103)
hamiltonian. However, many (most) solid state calculations do need and use relativistic corrections. This, in most cases, involves using a relativistic pseudopotential (an atomic necromancy problem) plus a spin-orbit operator. • Born-Oppenheimer approximation: the dynamic of electrons and nuclei can be approximately decoupled. Electrons move on a fixed nuclear frame. Nuclei move adiabatically over the electronic ground state potential energy surface. However, electron-phonon interaction is quite important: use perturbation theory afterwards. • Independent electron approximation: the many electron wave function is implicitely described as a Slater determinant over some spinorbital set. Correlation is treated through the ˆ Vxc functional. c V. Lua˜ na, QTC Murcia 2008 (104)
rough timeline 1928 Bloch theorem. 1931 Wilson: band theory shows the difference between insulators and metals. 1934 Slater: calculation of the bands of Na. 1935 Wigner and Seitz: first quantitative calculation of band structure in Na. 1935 Bardeen: Fermi surface of a metal. 1937 Slater: formulation of the APW method. 1940 Herring: formulation of the OPW method. 1947 Shockley-Bardden-Brattain: build a point contact transistor. 1947 Korringa (completed by Kohn and Rostoker in 1954): KKR method. 1951 Philips-Kleinman on pseudopotentials. 1953 Herrman-Callaway: first realistic band structure of a semiconductor (Ge). 1965 Hohenberg-Kohn theorem, and Kohn-Sham method. 1975 Anderson: LMTO method. Opens the door to the full potential ”L” methods. 1985 Car-Parrinello method. c V. Lua˜ na, QTC Murcia 2008 (106)
Free electron (1D) The Hamiltonian and the Schr¨ odinger equation: ˆ h = − 2m d2 dx2 = ˆ p2 x 2m , ˆ px = −i d dx . d2ψ dx2 = − 2m 2 ψ = −k2ψ =⇒ ψk(x) = eikx, = k2 2 2m , k ∈ R. (116) The solution is a 1D planewave (PW). Some properties: • PW(1D) are eigenfunctions of ˆ px: ˆ pxψk(x) = k ψk(x). • ψk(x) and ψ−k(x) have the same energy and move on opposite directions with the same speed. • A PW has an uniform density in R: ψk ψk = e−ikxeikx = e0 = 1 • A PW is not normalizable on R: R |ψk|2 dx = R dx −→ ∞. This should be evident from the fact that a PW is an eigenfunction of momentum: the position uncertainty should go to infinity through Heisenberg principle. • Spacial periodicity: Let x and x + λ be the closests different points in phase, i.e. ψk(x + λ) = ψk(x) for arbitrary x. Then eikxeikλ = eikx and eikλ = 1, so kλ = 2π. A PW has a wavelength λ = 2π/k . c V. Lua˜ na, QTC Murcia 2008 (108)
Periodic boundary conditions ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¢ If the PW is forced to be periodic with period A, i.e. if for any x ∈ R ψk(x + a) = ψk(x) =⇒ eika = 1 =⇒ ka = n2π, n ∈ N. (117) Only those PW with k being an integer multiple of 2π/a will have the desired periodicity. There still are an infinite number of PW, but we have passed from k ∈ R to n ∈ N. The PW can be normalized to a single cell of the 1D lattice: |N|2 a 0 |ψk|2 dx = 1. The set of normalized an periodic planewaves is |k = ψk(x) = 1 √ a eikx, k = n 2π a , k = k2 2 2m = h2n2 2ma2 , n ∈ N. (118) Any two different PWs from this set are orthogonal: k |k = 1 a a 0 ei(k−k )xdx = 1 a ei(k−k )a − 1 k − k = ei2π(n−n ) − 1 2π(n−n ) = 0 iff n = n . (119) The periodic PWs form, in fact, a complete set and, for any function 1 a k eik(x−x ) = δ(x−x ) =⇒ f(x) = k fkeikx ⇐⇒ fk = ∞ −∞ f(x)e−ikxdx. (120) f(x) is the Fourier transform of fk, and fk is the inverse Fourier transform of f(x). c V. Lua˜ na, QTC Murcia 2008 (109)
Free electron (3D) The hamiltonian is ˆ h = ˆ p2/2m, ˆ p = −i ∇, and the solutions are planewaves (PW) again: |k = ψ k (r ) = V −1/2eik·r, k = 2k2 2m , r, k ∈ R3. (121) with V being the volume of the normalization box. Some properties • The PWs are eigenfunctions of the momentum operator: ˆ p |k = k |k . • The particle velocity is proportional to k, the wavevector : v = k/m. • The |k and |−k PWs are degenerated. • The wavelength of a PW is λ = 2π/k. To enforce periodic boundary conditions in a general way we define the parallelepipedic cell a ˜ and an arbitrary primitive translation t = a ˜ n, n ∈ N3. For any r ∈ R3: ψ k (r + t ) = ψ k (r ) =⇒ eik·t = 1. (122) If k is a vector in the reciprocal cell, k = a ˜ k = 2πa ˜ h, the periodicity condition is 1 = eik·t = ei2π(h Tn) =⇒ h1nx + h2ny + h3nz ∈ N for all n ∈ N3. (123) c V. Lua˜ na, QTC Murcia 2008 (110)
¡ ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ ¢£ The wavevector of the periodic PWs is then k = 2π(h1a +h2b +h3c ) = 2πa ˜ h = a ˜ k, (h1, h2, h3) ∈ N3. (124) This discrete mesh of allowed k-points is distributed uniformly in the re- ciprocal space. Each k point can be associated with a small parallelepiped of volume v k = (2π)3V = (2π)3/V , where V is the volume of the main cell. It is implicitely assumed that a primitive cell is used to describe the reciprocal space. The periodic PWs form an orthonormal set k|k = 1 V V ei(k−k )rdr = δ k,k , (125) where the integral is done on a unit cell. The set is also complete, which means that any 3D function can be expanded as f(r ) = k f k eik·r ⇐⇒ f k = V f(r )e−ik·rdr. (126) c V. Lua˜ na, QTC Murcia 2008 (111)
Model: A gas of free and independent electrons Let’s assume a very simple model for a metal. Ne electrons are confined within the metal obeying the crystal lattice periodicity and the Pauli principle but independent and suffering no interaction otherwise. Each electron can be described using a PW plus a spin function: |kσ , where σ = ±1/2 is the spin-azimuthal quantum number. Let n k,σ (either 0 or 1) be the occupation number of spin-PW |kσ . A microscopic description of the state of the whole gas of electrons is obtained by giving the occupancies of all spin-PWs: {n k,σ }. The total number of electrons and the total energy of the gas is Ne = kσ n kσ , E = kσ n kσ k , (127) where k = 2k2/2m is the energy of PW |k . Fermi energy/sphere/radius: In the ground state of the gas, all energy levels from zero up to some energy F are occupied without gaps. The occupied states form a sphere in k-space. The volume of the sphere must be enough to accomodate the Ne/2 electron pairs, taking into account that each state occupies a volume of (2π)3/V : 4 3 πk3 F = Ne 2 (2π)3 V =⇒ kF = 3π2 Ne V 1/3 =⇒ F = 2 2m 3π2 Ne V 2/3 . (128) c V. Lua˜ na, QTC Murcia 2008 (112)
Example: Li bcc (a = 3,46 ˚ A for the I cell). Ne/V = 1/(a3/2) = 4,828 m−3 (each Li transfers one valence electron to the electron gas). Then: kF = 1,13 · 1010 m−1 = 1,13 ˚ A−1, F = 7,75 · 10−19 J = 4,84 eV. Those values are typical of many metals. Density of states: Allowed states form a discrete mesh in k-space. Their number is, however, so huge that no significant error is made by assuming that they form a continuous distribution. If n = N/V = k3/3π2 is the density of electrons with wavevector modulus ≤ k: dn = k2 π2 dk = g(k)dk = 1 2π2 2m 2 3/2 √ d = g( )d (129) where k and belong in the [0, ∞) interval. If we assume that all levels up to the Fermi energy are fully occupied, the total number of electrons and the total energy of the electron gas would be N V = F 0 g( )d , E V = F 0 g( )d . (130) Statistical treatment: Fermi-Dirac distribution. We can introduce the above energy distribution into an statistical treatment of the electron gas. The grand canonical partition function for a system of independent and identical fermions is Ξ(µ, V, T) = i (1 + λe−β i ) = i [1 + e−β( i−µ)] (131) where µ (chemical potential), V (volume), and T (absolute temperature) determine the thermody- c V. Lua˜ na, QTC Murcia 2008 (113)
namical state of the Fermi gas; β = 1/kBT; λ = eµβ = eµ/kBT is the Lewis-Randall activity of the gas; and the i runs over the one-particle quantum states, i.e. the |kσ states in the case of the electron gas. The average properties of the grand canonical ensamble are obtained from its partition function: pV = kBT ln Ξ, dE = −pdV + TdS + µdN, E = −pV + TS + Nµ, d(pV ) = pdV − SdT + Ndµ, =⇒ S = −kBT ∂ ln Ξ ∂T µ,V , N = kBT ∂ ln Ξ ∂µ T,V . (132) The average number of electrons is not a constant in the grand canonical esamble, but a function of the thermodynamic state. The average number of electrons per state follows the Fermi-Dirac distribution: N = i ni = kBT ∂ ∂µ i ln(1+e−β( i−µ)) ⇒ n( ) = 1 eβ( −µ) + 1 = fF D( /µ; βµ). (133) The Fermi-Dirac distribution is a universal function of two adimensional variables: /µ, the energy relative to the chemical potential; and βµ = µ/kBT, the reduced temperature. At low temperature µ ≈ F is a very good approximation. The Fermi temperature TF = F /kB is usually 1·103–1·104 K for most metals. Under room temperature T ≪ TF and the distribution is almost a step function. c V. Lua˜ na, QTC Murcia 2008 (114)
Derivative of the Fermi-Dirac function and integrals involving it. The function ∂f ∂ = − βeβ( −µ) eβ( −µ) + 1 2 (134) gets involved in many integrals related to the FD distribution. Most of the integrals lack a closed analytical form, but we can exploit the fact that ∂f∂ is close to a Dirac’s delta function to expand the integrals into a rapidly convergent series. The prototype integral is A = ∞ 0 F( ) ∂f ∂ d = η = β( −µ) dη = βd η ∈ [−βµ, ∞) = − ∞ −βµ F µ+ η β eη (eη + 1)2 dη ≈ − ∞ −∞ (135) We have seen that µ ≈ F kBT, in other words µ η/β. We can expand the F( ) function in a Taylor’s series: F µ+ η β = F(µ) + F (µ) η β + 1 2 F (µ) η β 2 + . . . (136) and using the auxiliary integrals Am = 1 m! ∞ −∞ ηmeηdη (eη + 1)2 (m ≥ 0) : A0 = 1, A1 = 0, A2 = π2/6, . . . (137) c V. Lua˜ na, QTC Murcia 2008 (117)
we get A = ∞ 0 F( ) ∂f ∂ d = −F(µ) − π2 6β F (µ) + . . . (138) Using this technique we can obtain: The average number of electrons: n = Ne V = 1 3π2 2m 2 3/2 µ3/2 1 + π2 8µ2β + . . . The chemical potential: µ = F 1 − π2 12 kBT F 2 + . . . The internal energy: Eel V = 1 5π2 2m 2 3/2 5/2 F 1 + 5π2 12 2 F β2 + . . . The cv heat capacity: cel v = k2 B 6 2m 2 3/2 √ F T If we add up the vibrational contribution, the heat capacity at low temperatures for a nonmagnetic metal is cv = α1T + α2T3. The lineal term comes from the conducting electrons. The cubic term is due to the elastic vibrations. c V. Lua˜ na, QTC Murcia 2008 (118)
Brillouin zones The first Brillouin zone (BZ-1) is the common name for the Wigner-Seitz primitive cell of the reciprocal or k-space lattice. Whereas primitive and centered cells are both used for the direct lattice, centering is avoiding in the manipulation of the k lattice. An alternative definition for BZ-1 is the set of points in k space that can be reached from the origin (k = 0) without crossing any Bragg plane. A Bragg plane for any two points in the lattice being the plane which is perpendicular to the line between the two points and passes through the bisector of that line. The concept of BZ-1 can be generalized. The second BZ (BZ-2) is the set of points that can be reached from the first zone by crossing only one Bragg plane. BZ-(n+1) is formed is the set of points not in {BZ-1, BZ-2, ... BZ-(n−1)} that can be reached from BZ-n by crossing only one Bragg plane. Alternatively, the n Brillouin zone can be reached from the origin by crossing n−1 Bragg planes, but not fewer. The construction of the BZ is illustrated in the next slides for a simple square lattice. An important point to check is that every Brillouin zone has the same volume, namely the volume of a primitive reciprocal lattice. In addition, any BZ can be mapped back to the first zone by using just primitive translations, i.e. all the BZ’s are equivalent by translation symetry. c V. Lua˜ na, QTC Murcia 2008 (119)
BZs are equivalent due to the translational part of the crystal space group. Furthermore, the rotational symmetry determines the equivalence between different positions within a Brillouin zone. The special symmetry points receive particular names. Although there are different naming conventions, Γ is typically used to designate the origin of the reciprocal cell (k = 0). Let’s examine the BZ for the cubic P, I and F Bravais lattices, directly from the Kvec plots of the excellent Bilbao Crystallographic Server. The plot and the list of special k positions is available for all the space groups. Fm¯ 3m L z kz Γ kx ky W V Σ S 1 3 X Λ ∆ K M Q 1 X U X S Im¯ 3m k k x y z kz H G N Σ Γ ∆ D Λ F P F 1 R N 1 3 H F 2 Pm¯ 3m z kz Γ Λ ky ∆ Z kx X M Σ R T S 3 X c V. Lua˜ na, QTC Murcia 2008 (124)
Brillouin zones Mapping the Fermi surface onto the Brillouin zones At 0 K the electrons occupy spin states up to filling up some region of k-space (a sphere in the case of a free electron gas). The most important part is the boundary between the populated and the empty regions, i.e. the Fermi surface. This boundary will tipically extend along several BZs, but it can be mapped back to the first BZ by means of primitive translations. The next two examples correspond to a free electron gas on a square lattice with a density, Ne/V , of 1 and 2 electron pairs per primitive lattice, respectively. Even this simple system can show a rather involved surface. For temperatures above 0 K the electron population is governed by the Fermi-Dirac statistics, so the boundary between populated and unpopulated states is broken by the thermal excitation of the electrons. The Fermi surface remains a useful concept, however, because TF = F /kB is quite large in real materials. Thermal effects can be described as a broadening of the surface boundary. The Fermi surface, is useful for predicting the thermal, electrical, magnetic, and optical properties of metals, semimetals, and doped semiconductors. As we shall see, F for an insulator lies on an energy gap, and only conducting materials have a Fermi Surface. Fermi surfaces can be measured through observation of the oscillation of transport properties in magnetic fields. The most direct technique is the angle resolved photoemission spectroscopy (ARPES, see [32]), that resolves the electronic structure in the momentum-energy space. c V. Lua˜ na, QTC Murcia 2008 (125)
Brillouin zones Periodic Table of the Fermi Surfaces of Elemental Solids http://www.phys.ufl.edu/fermisurface Ferromagnets: Alternate Structures : Tat-Sang Choy, Jeffery Naset , Selman Hershfield, and Christopher Stanton Physics Department, University of Florida Seagate Technology Jian Chen Source of tight binding parameters (except for fcc Co ferromagnet): D.A. Papaconstantopoulos, Handbook of the band structure of elemental solids, Plenum 1986. This work is supported by NSF, AFOSR, Research Corporation, and a Sun Microsystems Academic Equipment Grant. (15 March, 2000) Co_fcc Co_fcc c V. Lua˜ na, QTC Murcia 2008 (128)
a periodic potential Independent electrons in a periodic potential. Bloch theorem. The Scr¨ odinger equation for an independent electron in a potential V (r) is ˆ h(r)ψ(r) = − 2 2me ∇2 r + V (r) ψ(r) = ψ(r) (139) and the potential has the lattice periodicity, V (r + L), where L is any direct lattice vector. As ∇r = ∇ r+L , because L is constant, it follows that ˆ h(r + L) = ˆ h(r). In the case of a non-degenerate energy level: Let’s assume ˆ h(r + L)ψ(r + L) = ˆ h(r)ψ(r + L) = ψ(r + L) and ˆ h(r)ψ(r) = ψ(r) (140) As ψ(r + L) and ψ(r) share the same non-degenerate energy, they must coincide up to a multiplier: ψ(r + L) = ζ(L)ψ(r). The multiplier ζ(L) must be such that • ζ(L) 2 = 1, if we ask that both ψ(r + L) and ψ(r) are normalized; • ζ(L + L ) = ζ(L)ζ(L ), because two succesive translations by L and L are equivalent to a single translation by L + L . To achieve this: ζ(L) = eiq − → L , where q can be any vector in k-space. Then: ψ(r + L) = eiq − → L ψ(r) (Bloch theorem). (141) c V. Lua˜ na, QTC Murcia 2008 (129)
a periodic potential In the case of a degenerate energy level: ψi(r + L) will be, in general, a linear combination of the degenerate states in r: ψi(r + L) = j ζij(L)ψj(r). (142) We can always ask that the degenerate functions form an orthonormal set: ψi|ψj = δij. The unitary character of the ζ matrix follows from here: δik = ψi(r + L)|ψk(r + L) = j,l ζ∗ ij (L)ζkl(L) ψj(r)|ψl(r) = j ζ∗ ij ζkj ⇒ ζ−1 = ζ†. (143) This means that we can diagonalize ζ and the eigenvectors will not mix under the translation L. From here we continue as in the non-degenerate case. Restrictions on q due to the lattice network in k-space: If G is a reciprocal lattice vector eiGL = 1. Then, for any q in k-space ei(q+G)L = eiGLeiqL = eiqL. In other words, q can be chosen to be within the first BZ. Periodic Boundary Conditions (Born-von Karman): Let’s assume the crystal to be formed by N = N1 × N2 × N3 parallelepipedic cells (along the a, b, c directions, respectively). By impossing equivalence of the one particle wavefunctions on the opposite faces we introduce quantization of c V. Lua˜ na, QTC Murcia 2008 (130)
a periodic potential the q vector. For instance, along the a direction ψq(r) ≡ ψq(r + N1a) = eiqN1aψq(r) ⇒ eiqN1a = 1 ⇒ q1N1 = i2π (144) The q1 component can get N1 independent values: 2π/N1, 4π/N1, . . . , 2N1π/N1. Equivalence at the boundaries along b and c introduce the quantization of the components q2 and q3. The N1 × N2 × N3 independent values for the q vector reside in the first BZ and have equivalent copies on each and every BZ. As N is usually quite large (≈ NA), q can be treated as a continuous variable for most purposes. Each independent q labels a one particle orbital that can be occupied by a pair of electrons of different spin. The ground state of a crystal with n electron pairs per primitive unit cell will consist of n different sets of ψnq(r), with energies nq , populated by the electron pairs. Thermal effects will introduce some smearing of the electron population along the states close to the Fermi surface. Bloch functions: Felix Bloch demonstrated in 1928 that one electron wavefunctions in a periodic potential can be described as the product of an arbitrary planewave and a function, called envelope, that satisfies the periodicity of the crystal. In other words ψnq(r) = eiqrunq(r) with unq(r+L) = unq(r) (145) where q ∈ R3 is any vector in k-space, L is a lattice vector in direct space, and r is an arbitrary c V. Lua˜ na, QTC Murcia 2008 (131)
a periodic potential vector, also in direct space. The ψq(r) wavefunctions are called Bloch functions or Bloch states. This equation is different but equivalent to the Bloch theorem seen before. For a Bloch function: ψnq(r+L) = eiq(r+L)unq(r+L) = eiqLeiqrunq(r) = eiqLψnq(r) (146) which is the first form of the theorem (eq. 141). Inversely, for any function φq(r) satisfying 141 we can build a Bloch state using uq(r) = φq(r)/eiqr. Bloch states are delocalized on the whole crystal, showing identical shape on every single unit cell. On the contrary, they are localized in k-space, as q has a definite value (and we need only to consider q in the first BZ). Wannier functions: An equivalent but different way of describing the electronic states is using one particle wavefunctions that are localized in r-space but delocalized in k-space. This Wannier functions are related to the Bloch states as: ψnq(r) = 1 √ N L eiqLWn(r−L) or Wn(r−L) = 1 √ N q∈BZ1 e−iqLψnq(r) (147) c V. Lua˜ na, QTC Murcia 2008 (132)
schemes Mapping the energy states onto the Brillouin zones. Energy bands. The representation of the energy levels nq ≡ n(q) as a function of q gives rise to the band structure diagram, perhaps the most typical outcome of band theory. To avoid dealing with a 4D map, it is customary to define a trajectory in k-space that follows the directions of maximal symmetry on the reciprocal cell. The n(q) values are then ploted versus the length of the trajectory from 0 to q. The band structure diagram can be limited to the first Brillouin Zone (reduced sone scheme), because all BZ are equivalent by the translational symmetry. The n band can also be represented in the n BZ (extended zone scheme) and this is the origin of a very simple recipe to use the free electron model as a method of drawing a first approximation to the band structure diagram: • Each reciprocal lattice point, G, is considered the source of a set of planewaves. Each G will produce a band within the first BZ, i.e. n ≡ G. • The energy at q of the planewave originated at G is n≡G (q) = 2 q − G 2 /2m. • In the reduced zone scheme we only need to sample q ∈ BZ1 and G runs over the whole lattice. In the extended zone scheme we only use G = 0 but q samples all BZ’s. • Bands will show degeneracy at the boundaries of the BZ’s. This can be avoided with a simple perturbation theory treatment. (PW’s interfere for the Bragg planes) c V. Lua˜ na, QTC Murcia 2008 (133)
schemes Γ ∆ Ζ Μ Σ Χ The red band origi- nates at the (0, 0) k-point, green bands come from (1, 0) and equiva- lents, blue bands from (1, 1), and magenta ones from (2, 0). 0.0 1.0 2.0 3.0 4.0 5.0 Γ ∆ X Z M Σ Γ ∆ X ε / (h2/2me a2) c V. Lua˜ na, QTC Murcia 2008 (137)
model The Nearly Free Electron Model (NFEM) A simple way to improve on the free electron bands is by treating the collective effect of the rest of the crystal as a little perturbation on top of the free electron hamiltonian. Orthonormal PWs are just the solution for the unperturbed problem: ˆ H(0) = − 2 2m ∇2 r , ψ(0) k (r) = |k = V −1/2eikr, (0) k = 2k2 2m , k = 2πa ˜ h, h ∈ Z3. (148) Let ˆ V (r), the lattice potential on an electron, be treated as a first order perturbation. Wave functions and eigenvalues expanded in a perturbation series, ψ k (r) = ψ(0) k + ψ(1) k + ψ(2) k + . . . , k = (0) k + (1) k + (2) k + . . . , (149) can be substituted into the one-particle Schr¨ odinger equation, ( ˆ H(0) + ˆ V )ψ k = k ψ k . (150) Grouping together the terms with the same order in the perturbation we get zero order: 0 = ( ˆ H(0) − (0) k )ψ(0) k ; (151) first order: 0 = ( ˆ H(0) − (0) k )ψ(1) k + ( ˆ V − (1) k )ψ(0) k ; (152) second order: 0 = ( ˆ H(0) − (0) k )ψ(2) k + ( ˆ V − (1) k )ψ(1) k + (− (2) k )ψ(0) k ; . . . (153) c V. Lua˜ na, QTC Murcia 2008 (138)
model The zero order equation is just the unperturbed problem. To solve the first order equation we expand ψ(1) k in PW series ψ(1) k (r) = k =k c(1) kk ψ(0) k (r). (154) This expansion is always possible because PWs for a complete set. Substituting 154 into 152 and multiplying by ψ(0) q | 0 = k =k c(1) kk ψ(0) q |( ˆ H(0) − (0) k )|ψ(0) k + ψ(0) q |( ˆ V − (1) k )|ψ(0) k = k =k c(1) kk ( (0) k − (0) k ) ψ(0) q |ψ(0) k + ψ(0) q | ˆ V |ψ(0) k − (1) k ψ(0) q |ψ(0) k = k =k c(1) kk ( (0) k − (0) k )δ q,k + ψ(0) q | ˆ V |ψ(0) k − (1) k δ q,k = c(1) kq ( (0) q − (0) k ) + ψ(0) q | ˆ V |ψ(0) k − (1) k δ q,k (155) where we have used the orthonormality of the lattice PWs. In fact, we have used 1 V V eiGrdr = 0 iff G = 2πa ˜ n, n ∈ Z3 (156) where V is the direct lattice cell. c V. Lua˜ na, QTC Murcia 2008 (139)
model The integral of the perturbation is very important. Using a Fourier series expansion of the potential: V (r) = g Vgeigr (157) k| ˆ V |k = g Vg k|eigr|k = g Vg k|k +g = g Vgδ k,k +g = V k−k (158) Notice: (1) The Fourier series of V (r) sums only over reciprocal lattice vectors, due to the periodic nature of the potential; (2) The integral holds true whenever k−k is a reciprocal lattice vector, even if k and k are not. Using 158 in 155 we arrive to 0 = c(1) kq ( (0) q − (0) k ) + V q−k − (1) k δ q,k (159) and Case q = k: (1) k = V 0 ; Case q = k: c(1) kq = − V q−k (0) q − (0) k . (160) All energy levels are increased by the value of the lattice potential at the Γ k-point. The zero of the energy scale is arbitrary, however, and we can set V 0 = 0. Up to first order, then, the lattice potential adds nothing to the free electron band structure and we need to proceed to second order, at least. c V. Lua˜ na, QTC Murcia 2008 (140)
model Obtaining the second order correction is similar to the first order already done. The ψ(2) k wave function can also be expanded in PW series ψ(2) k (r) = k =k c(2) kk ψ(0) k (r). (161) Using 161 and 157 in 153 and multiplying by ψ(0) q |: 0 = k =k c(2) kk ( (0) k − (0) k ) ψ(0) q |ψ(0) k + k =k c(1) kk ψ(0) q | ˆ V |ψ(0) k − (2) k ψ(0) q |ψ(0) k = k =k c(2) kk ( (0) k − (0) k )δ q,k + k =k c(1) kk V q−k − (2) k δ q,k = c(2) kq ( (0) q − (0) k ) − k =k V k −k V k−k (0) k − (0) k − (2) k δ q,k . (162) The second order corrections to energy and wave functions are then For q = k: (2) k = − k =k |V k−k |2 (0) k − (0) k ; For q = k: c(2) kq = k =k V k −k V q−k ( (0) k − (0) k )( (0) q − (0) k ) ; (163) where we have used that V−g = V g because V (r) is a real function. c V. Lua˜ na, QTC Murcia 2008 (141)
model All the derivation above fails when we try to get the perturbative corrections to a degenerated state. This is, however, a most important situation, as degeneracy occurs systematically at the BZ boundaries. We can treat the problem using perturbation theory for degenerate states. Let |ψ(0) k and |ψ(0) k+q be two almost degenerated zero order states. The first order correction to the energy is obtained by solving the secular equation: 0 = (0) k − ψ(0) k | ˆ V |ψ(0) k+q ψ(0) k+q | ˆ V |ψ(0) k (0) k+q − = (0) k − V q Vq (0) k+q − = 2 − ( (0) k − (0) k+q ) + (0) k (0) k+q − |Vq|2 (164) and = (0) k + (0) k+q 2 ± (0) k − (0) k+q 2 2 + |Vq|2 1/2 (165) In the degenerated limit (0) k = (0) k+q and = (0) k ± |Vq|. The figure in page 144 shows the corrections to the free electron bands of a one dimensional lattice calculated with eq. 163. It is clear that the formulas diverge in the neighborhood of a degeneracy. In fact, a 0/0 indeterminacy happens in that case. c V. Lua˜ na, QTC Murcia 2008 (142)
model Eq. 165, on the other hand, works like a charm as the figures in page 145 and 146 clearly show. The result is easier to describe in the extended or repeated zone schemes. The energy parabola typical of the free electrons curves up or down in the nearby of a BZ boundary. This generates gaps of forbidden energy, i.e. energy ranges for which no state occurs. The width of the gap is, in this model, twice the absolute value of the lattice potential at the BZ boundary. The reason of the success of the eq. 165 can be questioned. The derivation is purported for the degenerated case but the formula works well quite far from this situation. In fact, it works for any k-point and it does not suffer the problems of the second order perturbation formula, eq. 163. Even though solid state physics books systematically describe the nearly free electron model in terms of perturbation theory, it is better to analyze the problem as a variational linear calculation. The complete, albeit approximated, hamiltonian is ˆ H = ˆ T + ˆ V (r), and the PWs are the basis set functions. The Schr¨ odinger equation is transformed into a set of secular equations: det |H − k 1| = 0 and (H − k 1)c k = 0 (166) where the H matrix elements are H k,k = ψ(0) k | ˆ T + ˆ V |ψ(0) k = T k,k + V k,k , T k,k = (0) k δ k,k , V k,k = V k−k . (167) A basis set formed by the two PWs |k and |k+q will give rise to the eq. 165. The variational method, however, can be used with a basis of any size, the only trouble being the use of an efficient method for diagonalizing the H matrix. c V. Lua˜ na, QTC Murcia 2008 (143)
Abinit: GPL, www.abinit.org, X. Gonze et al. (Universit´ e Catholique de Louvain; Corning Inc.; etc). Pwscf: GPL, www.pwscf.org, S. Baroni et al. (Trieste, . . . ). Wien2k: distributed as source for a small subscription, www.wien2k.at, P. Blaha (K. Schwartz) et al. (Technical University Wien, . . . ) Siesta (Spanish Initiative for Electronic Simulations with Thousands of Atoms): distributed as source for a small subscription, www.uam.es/siesta/, E. Artacho (U. Cambridge), P. Ordej´ on (ICM, Barcelona), J. Junquera (U. Cantabria), D. S´ anchez Portal (UPV), J. M. Soler (UAM), . . . et al. crystal06: only binary is distributed (06) or source could not be modified (03 and 98 versions), www.crystal.unito.it, R. Dovesi (U. Torino), V.R. Saunders (Daresbury Lab., U. Cambridge) et al. References Kantorovich, 2004 [33]; Kaxiras, 2003 [34]; Kittel, 2005 [35]; Marder, 2000 [36]; Martin, 2004 [37]; McQuarrie, 1976 [38]; Pueyo, 2005 [39]; See Damascelli et al. [32] for a description of the Fermi Surface measurement of high-Tc cuprate c V. Lua˜ na, QTC Murcia 2008 (147)
1. Demonstrate the next properties of planewaves: (a) the wavelength of the ψ k (r) planewave is λ = 2π/k; (b) periodic PWs form an orthonormal set; 2. The image in page 110 sketches a direct and its corresponding reciprocal lattice. The image is, however, wrong. Can you point the mistakes included and plot schematically the correct reciprocal lattice? 3. Determine F for a free electron gas in a simple square lattice if there are three electron pairs per unit cell. Use the corresponding Fermi radius to map the Fermi sphere over the Brillouin zone scheme, and plot the shape of the Fermi surface once translated to the first Brillouin zone. 4. Determine and plot the band structure diagram for a free electron in a simple square lattice. The special points in the first Brillouin zone are (k-coordinates in 2π/a units): Γ(0, 0), X(1/2, 0), and M(1/2, 1/2). The special lines are ∆ (from Γ to X), Σ (Γ-M) and Z (X–M). 5. c V. Lua˜ na, QTC Murcia 2008 (148)
The free electron model totally ignores the nature of the material apart from the crystal geometry and the assumption on the number of electrons donated, per unit cell, to the electron gas. The nearly free electron model introduces a dependency on the lattice potential while retaining the concept that the valence electrons belong to the whole crystal. The tight-binding model, on the other hand, is based under the assumption that the electronic structure of the solid very much resembles that of their atomic components. Crystalline orbitals are thus derived from the atomic orbitals by imposing on them the periodicity required by Bloch’s theorem. Albeit the TB scheme can be applied ab initio, it is common to introduce great simplifications to produce final TB formulas depending upon a short collection of parameters, empirically fitted to some key experimental data. Many TB models can be linked back to the influential article by Slater and Koster [40]. A good detailed description of the TB calculations is found in chapter 4 of Kaxiras [34]. A recent review of TB techniques and applications can be found in Goringe, Bowler and Hern´ andez, 1997 [41]. Let φl(r − ti) be the l-th atomic orbital attached to atom i, whose position is ti with respect to the origin of the unit cell in which it is placed. Bloch states can be obtained as χ kil (r) = 1 √ N R eikRφl(r − ti − R) (168) where R sums over the N primitive unit cells that from the crystal. c V. Lua˜ na, QTC Murcia 2008 (150)
be expanded in the basis of the TB Bloch functions: ψ nk (r) = il c nkil χ kil (r) (169) where the sum runs on the atoms in the primitive unit cell (i) and the atomic orbitals (l). Substitution of this expansion in the Schr¨ odinger equation gives ˆ hψ nk (r) = nk ψ nk (r) =⇒ il c nkil ˆ h − nk χ kil (r) = 0. (170) Upon multiplication by χ kmj | il c nkil χ kmj |ˆ h|χ kil − nk χ kmj |χ kil = 0 (171) the differential equation is converted into a secular equation, on which the band energies, nk , and the band coefficients, c nkil , are the unknowns to solve. We can write the secular equations in terms of atomic orbitals. The overlap between two Bloch states is given by χ kmj |χ kil = 1 N R ,R eik(R −R ) φm(r − tj − R )|φl(r − ti − R ) (†) (172) = 1 N R,R eikR φm(r − tj)|φl(r − ti − R) = (‡) R eikR φm(r − tj)|φl(r − ti − R) (173) c V. Lua˜ na, QTC Murcia 2008 (151)
into account that (†) the atomic overlap integral should not change if we translate equally both atomic functions, R = R − R , and (‡) the term added up in 173 do not longer depend on R , so (1/N) R → 1. The hamiltonian integrals can be treated is a similar way to give χ kmj |ˆ h|χ kil = R eikR φm(r − tj)|ˆ h|φl(r − ti − R) . (174) The secular equation is then ∀ jm il c nkil R eikR φm(r−tj)|ˆ h|φl(r−ti−R) − nk φm(r−tj)|φl(r−ti−R) = 0 (175) and it has to be solved on each k point within the first BZ. So far, the TB equations could be solved ab initio. However, most of the TB calculations introduce severe simplifications to let the equations be solved ”by hand” and even produce analytical expressions for the band energies. Let us examine one of the most common schemes. First, atomic orbitals on different centers and different orbitals on the same center are assumed to be orthogonal φm(r−tj)|φl(r−ti−R) = δlmδijδ(R). (176) All hamiltonian elements are asumed to be negligible except the orbital energy of an atomic orbital on a single center (on site energies, el) and the integrals between atomic orbitals on nearest c V. Lua˜ na, QTC Murcia 2008 (152)
Vlm.ij): φm(r−tj)|ˆ h|φl(r−ti−R) = elδlmδijδ(R), = Vlm,ijδ((tj−ti − R) − dnn), (177) where dnn is a vector between the i-th atom and its nearest neighbors. Example: 1D linear chain ¡ ¡ ¡ ¡ ¡ ¡ ¡ ¢ Let x be the direction aligned with the chain. The Bloch funcions are χkl(x) = 1 √ N n∈Z eiknaφl(x − na) (178) where l is the type of atomic orbital (s, pσ = px and pπ = {py, pz}); N is the number of primitive unit cells, i.e. the number of n values. In the simplified TB scheme: Overlap: φm(x−ia)|φl(x−[i+n]a) = φm(x)|φl(x−na) = δlmδn0; (179) Hamilton: φm(x−ia)|ˆ h|φl(x−[i+n]a) = ... = = elδlmδn0; = Vlmδn,±1. (180) c V. Lua˜ na, QTC Murcia 2008 (153)
we need the next hopping integrals Vppπ Vppσ Vspσ Vssσ (181) whereas the next cases are trivially null due to the symmetry (182) The Schr¨ odinger equation takes the form of a secular equation ∀ m : 0 = l ckl n∈Z eikna elδlmδn0+Vlmδn,±1 φm(x)|ˆ h|φl(x−na) − k δlmδn0 φm(x)|φl(x−na) = l ckl (el − k)δlm + [eika + e−ika] 2 cos(ka) Vlm (183) c V. Lua˜ na, QTC Murcia 2008 (154)
notation defining: Vklm = 2Vlm cos(ka) and Ekl = el + Vkll. Then Eks − k Vkspσ 0 0 Vkspσ Ekpσ − k 0 0 0 0 Ekpπ − k 0 0 0 0 Ekpπ − k cks ckpσ ckpy ckpz = M k ck = 0 (184) and the secular equation is clearly separated into a 2 × 2 and two 1 × 1 independent blocks. The eigenvalues for the σ bands are 0 = Eks − k Vkspσ Vkspσ Ekpσ − k ⇒ k = Eks+Ekpσ 2 ± Eks−Ekpσ 2 2 + V2 kspσ (185) and the energies of the two degenerated π bands are k = Ekpπ = ep + 2Vppπ cos(ka). (186) There is a secular system for each k-point in BZ1, i.e. k ∈ [− π a , + π a ]. The s and pσ functions mix together to form two σ orbitals. The σ and π functions do not mix in this system due to the symmetry. c V. Lua˜ na, QTC Murcia 2008 (155)
λ-type orbitals only. There is a sin- gle band, with energy k = Ekλ = eλ + 2Vλλ cos(ka). The band center is eλ, the atomic energy of the λ level, and its width is 2Vλλ, i.e. the witdh is determined by the hopping integral between two λ functions in nearest neigh- bor atoms. The atomic levels follow the aufbau principle, so we can expect a similar ordering for the crystal bands: 1s < 2s < 2p < 3s.... Assuming that Vssσ < 0, the s band shape is similar to the nearly free electron fundamental band. The ssσ hopping integral can be seen as +| − |+ , where the signs show the dominant component on each orbital lobe and on the operator, but then, for the ppσ hopping integral we have ±| − |∓ , and ±| − |± for the ppπ. We expect, then, Vppσ > 0 and Vppπ < 0. The figure on the side shows the close relationship between the ssσ, ppσ and ppπ bands with the first three bands in the free electron model. This striking relationship has been exploited by Harrison and oth- ers to determine the TB parameters that best fit the free electron bands [42]. π/2 0 -π/2 εk ka s band pσ band pπ band c V. Lua˜ na, QTC Murcia 2008 (156)
group IV diamond structures Harrison and coworkers [42, 43] have studied systematically the application of TB models to covalent, ionic and metallic solids. The next table contains all the data required to do a simple TB calculation on the diamond structure of C, Si, and Ge, prototypes of covalent solids with tetrahedral coordination. The calculation uses a minimal sp3 atomic basis set to describe the four valence electrons per atom, i.e. the eight valence electrons per primitive unit cell. C Si Ge a (˚ A) 3.57 5.43 5.66 s (eV) -17.52 -13.55 -14.38 p (eV) -8.97 -6.52 -6.36 Vssσ (eV) -4.50 -1.93 -1.79 Vspσ (eV) 5.91 2.54 2.36 Vppσ(eV) 10.41 4.47 4.15 Vppπ(eV) -2.60 -1.12 -1.04 Hopping integrals strongly depend on the interatomic distance. Harrison used a bond orbital model to obtain Vlm = 2 me ηlm d2 , 2 me = 1 at.u. = 7,62 eV ˚ A2, (187) where ηlm is a dimensionless constant and d is the nearest neighbors distance. This equation works ex- tremely well on tetrahedral semiconductors, but it is rather poor on, e.g., close-packed structures. Obtaining the TB equations and developing a simple code for this case is not difficult. We can also resort to the TBPW provided by R. M. Martin and cols. at the www.electronicstructure.org website. c V. Lua˜ na, QTC Murcia 2008 (157)
0 5 10 15 20 25 Γ U X Γ L C εk (eV) -10 -5 0 5 Γ U X Γ L Si -10 -5 0 5 Γ U X Γ L Ge Band energies have been dis- placed to make zero the top of the valence band. The three solids are predicted to have a direct band gap at Γ: 13,89 (C), 3,66 (Si), and 1,90 eV (Ge). C is predicted to be an insulator, and Si and Ge are predicted to be semicon- ductors. Experimental values for the direct band gap are 7,3 (C), 3,48 (Si) and 0,81 eV [44]. The three solids, how- ever, have a significantly lower indirect band gap: 5,48 (C), 1,11 (Si), and 0,66 eV (Ge). The sp3 minimal basis is able to fit well the valence bands, but it fails to describe the con- duction band structure. c V. Lua˜ na, QTC Murcia 2008 (158)
Γ U X Γ L Harrison Si εk (eV) -10 -5 0 5 Γ U X Γ L Vogl Vogl et al. [45] used a sp3s∗ basis to produce a very suc- cessful model of the tetra- hedral semiconductors. The s∗ excited state very much improves the fitting of the conduction band that now predicts correctly the indi- rect band gap: 1,17 eV on Si (Exptal.: 1,11 eV). Vogl paper describes a general method for fitting to high- symmetry points on the band structure. c V. Lua˜ na, QTC Murcia 2008 (159)
recipe The TB ideas can be carried up to a self-consistent ab initio procedure. Failing to reach that goal, let us present some common improvements over the basic TB recipe. Arbitrary orientation of the orbitals: The best strategy to deal with an arbitrary crystal is to assume a global set of cartesian axes. Given two atoms and a separation vector d/d = xi+yj+zk = sen θ cos φi+sen θ sen φj +cos θk, some of the non-null hopping integrals are (notice the illustration in next page): Vss=Vssσ; Vs,px =x2Vspσ; Vpx,px =x2Vppσ+(1−x2)Vppπ; Vpx,pz =xz(Vppσ−Vppπ). (188) Slater and Koster [40] provide all the formulas required to work with s, p and d orbitals. Nonorthogonality of the orbitals: The simplified TB equations tend to assume that atomic orbitals centered of different centers are orthogonal but they are not, in general. The overlap depends strongly on the internuclear distance and on the space extension of the particular orbitals: the average radius is proportional to the main quantum number, so orbitals that belong to the same shell have a similar space range. Taking into account the lack of orthogonality becomes unavoidable when the TB models go beyond the minimal basis formulation. Multicenter integrals: Three and four center integrals between exponential type orbitals are usually neglected, mostly because they are difficult and expensive to compute. Nowadays, there exist methods to calculate all of them with any required precision. c V. Lua˜ na, QTC Murcia 2008 (160)
addition of low lying excited state orbitals to the basis set can improve greatly the behavior of the TB equations. Vogl et al. [45] provide a classical example for the important role of the s∗ on the description of the conduction band of the tetrahedral C–Ge semiconductors. As a matter of fact, the TB method tends towards an ab initio technique as the number and flexibility of basis set increases well beyond the minimal basis range of the basical scheme. c V. Lua˜ na, QTC Murcia 2008 (161)
band structure • The atomic orbitals give rise to the bands in the solid. The band width is mainly consequence of the mixing of equivalent orbitals from nearby atoms, henceforth increasing as the hopping integrals increase. • Bands are filled by paired electrons in order of increasing energy. The highest occupied band is called valence band, whereas the conduction band is the lowest unnoccupied one. • Bands can be separated by gaps of forbidden energy. The most important one being the gap between the top of the valence band and the bottom of the conduction band. The gap is called direct when it involves valence and conduction states with the same k value, and indirect otherwise. • In absence of vibrations, the only spectroscopic transitions allowed by the main electric dipole mechanism must conserve the k value, i.e. they are vertical transitions in the band diagram. However, k → k transitions become allowed if a crystal vibration (phonon) of appropriate symmetry carries away the momentum excess (k −k). • Systems are classified according to the band gap energy Eg into: metals and semimetals (Eg = 0), semiconductors (0 < Eg < 3 ∼ 4 eV), and insulators (Eg 3 eV). Metals are good electrical conductors, semiconductors have an electrical resistivity lying in the range of 1 · 10−2–1 · 109 Ω cm, and the resistivity of insulators is much higher. • The electrical conductivity is related to the number of free carriers: electrons and holes in c V. Lua˜ na, QTC Murcia 2008 (162)
therefore able to pass to a electronic state of different momentum with a negligible energy cost. A semiconductor lacks free carriers at the absolute zero, but an increasing number of electrons can jump the energy gap as the temperature increases. The conductivity of a semiconductor, thus, increases with T. • Contrarily, the probability of carriers-nuclei collisions grow with temperature and this effect dominates the conductivity of good metals, that become less conductive when heated. c V. Lua˜ na, QTC Murcia 2008 (163)
sampling and van Hove singularities A quite useful concept in analyzing the electronic band structure of solids is the density of states (DOS) as a function of the energy. The g(ε)dε distribution represents the number of electronic states available in the energy range from ε to ε+dε. In the case of a free electron gas, we have already seen (eq. 129 in page 113) that g(ε) = 1 2π2 2me 2 3/2 √ ε (189) and the DOS follows a smooth g(ε) ∝ √ ε curve. Real solids, however, show a far more complex DOS. Taking into account the spin degeneracy and normalizing g(ε) to the volume of the solid: g(ε) = V (2π)3 nk 2δ(ε − nk ) = 2V (2π)3 n 2δ(ε − nk )dk = 2V (2π)3 n nk =ε dS k ∇ k nk (190) The last integral (see, e.g. Kaxiras [34], chap. 5) is over surfaces in k-space on which the band energy nk is constant, and dS k is the differential element normal to the surface. No matter how complex it looks, the last integral makes clear a very important feature: the DOS has sharp spikes called van Hove singularities [46] at the points such that ∇ k nk = 0, i.e. on the critical points of the energy bands in k-space. c V. Lua˜ na, QTC Murcia 2008 (164)
0.0 0.2 0.4 0.6 0.8 1.0 1.2 (ε-εF ) (eV) g(ε) (e/eV) Γ M K Γ A DOS of hcp Be, according to a FPLAPW GGA calculation [50]. c V. Lua˜ na, QTC Murcia 2008 (166)
[42]; Harrison, 2005 [43]; Kantorovich, 2004 [33]; Kaxiras, 2003 [34]; Kittel, 2005 [35]; Marder, 2000 [36]; Martin, 2004 [37]; Yu and Cardona, 2001 [44]. Exercises 1. Show that the χ kil (r) defined in eq. 168 satisfy Bloch’s theorem. 2. Consider a 2D square lattice with a single type of atoms located at the lattice grid points. Derive the TB equations for a system of orthogonal s and p orbitals. 3. Extend the above model to a 3D cube lattice. 4. Use the TBPW by R. M. Martin et al., or any other similar TB code, to determine the band structure of C, Si and Ge using the sp3s parametrization of Vogl et al. [45]. 5. Derive the DOS for the free electron model in one and two dimensions. 6. Calculate the DOS for the TB model of a 2D square lattice with one atom per unit cell and s and p orbitals. For this calculation it is not sufficient to obtain the bands along some high symmetry lines, but the entire BZ must be sampled with enough resolution to be able to show the characteristic patterns around the critical points. c V. Lua˜ na, QTC Murcia 2008 (167)
The independent electron Schr¨ odinger equation We want to solve the Hartree-Fock or Kohn-Sham equation for an electron in the average field of all other (infinite) crystal charges: ˆ H(r)ψnq(r) = − 2 2me ∇2 r + ˆ V (r, [ρ]) ψnq(r) = n(q)ψnq(r) (191) where ˆ V (r, [ρ]) = ˆ Vn + ˆ VH + ˆ Vxc : • ˆ Vn: electrostatic potential of the nuclei (or pseudopotentials of the chemical kernels); • ˆ VH (r) = R3 ρ(r )dr |r − r | : Coulomb potential due to the (possibly valence) electron density. • ˆ Vxc: Exchange and correlation term. Common approaches: Hartree-Fock (HF), LDA or LSDA (Local [Spin] Density Approach), GGA (Generalized Gradient Approach), . . . Notation: vectors on the direct and reciprocal spaces. r-space = {r = a ˜ x | x ∈ R3}; r-lattice = {L = a ˜ n | n ∈ Z3}; (192) k-space = {q = 2πa ˜ q | q ∈ R3}; k-lattice, = {G = 2πa ˜ n | n ∈ Z3}. (193) c V. Lua˜ na, QTC Murcia 2008 (169)
waves Electronic band structure via plane waves We will transform Schr¨ odinger equation to a secular form by expanding the crystal orbitals in a basis of planewaves. We start by describing a crystal orbital as a Bloch function: ψnq(r) = eiqrunq(r) = eiqr G cnq 1 √ V eiGr = G cnq 1 √ V ei(G+q)r = G cnq |G+q (194) where we have used the fact that the periodic component of the Bloch function, unq(r) = unq(r+L), can be expanded in series of orthonormal planewaves. The result is that the planewaves that form the basis of the crystal orbitals have the form |q+G , where G is a vector in the k-lattice and q is the same k-space vector that labels the crystal wavefunction that we want to expand. Using 194 in 191: 0 = ˆ H − n(q) ψnq(r) = G1 cnq(G1) ˆ H − n(q) |q+G1 (195) and multiplying by q+G2| 0 = G1 cnq(G1) q+G2| ˆ H|q+G1 − n(q) q+G2|q+G1 (196) c V. Lua˜ na, QTC Murcia 2008 (170)
waves but q+G2|q+G1 = G2|G1 = δ(G1 − G2) (197) because both G1 and G2 belong to the k-lattice, so |G1 and |G2 belong to the orthonormal PW set. The hamiltonian element matrix is a sum of the kinetic and potential energy components. The kinetic energy is trivial, as PWs are eigenfunctions of ˆ p and ˆ T = ˆ p2/2m: T G1,G2 (q) = q+G2| − 2 2m ∇2 r |q+G1 = − 2(q+G1)2 2m δ(G1 − G2). (198) The potential energy integral is greatly simplified if, taking into account the ˆ V (r+L) = ˆ V (r) lattice periodicity, we expand ˆ V (r) into a Fourier series: V G1,G2 (q) = G V G q+G2|eiGr|q+G1 = G V G δ(G1+G−G2) = V G2−G1 . (199) The potential energy is thus reduced to the calculation of a single coefficient in the Fourier series: Vg = V ˆ V (r)e−igrdr (200) where the integral is done within the volume, V , of a single unit cell. c V. Lua˜ na, QTC Murcia 2008 (171)
waves Using 198 and 199 in 196 we obtain the secular equation 0 = G1 cnq(G1) 2(q+G1)2 2m − n(q) δ(G1−G2) + V G2−G1 ∀ G2 (201) that must be solved independently for each q that belongs to the first Brillouin Zone (BZ1). The normalized crystal orbitals are then ψnq(r) = G cnq(G) |q+G with 1 = ψnq(r)|ψnq(r) = G cnq(G) 2 . (202) The electron density of the crystal is obtained as ρ(r) = nq fnq ψnq(r) 2 (203) where fnq is the electron population on band nq. In the case of a metal, this requires a delicate calculation of the Fermi energy. The electron density is a required component in the calculation of the Vg Fourier coefficients, because the V (r) lattice potential containts the Coulomb and the exchange-correlation contributions. Henceforth, the secular equations 201 must be solved self-consistently on each q-point in the BZ1. Obtaining the total energy, like the Density of States (DOS) and related properties, is a last step in the calculation, and it requires sampling efficiently the BZ1. c V. Lua˜ na, QTC Murcia 2008 (172)
waves The main difficulty for the scheme described is the number of planewaves required to describe any material. The electron density is heavily concentrated around the nuclei and, in fact, this is the region that contributes most to the total energy. Planewaves are quite inefficient to represent a tightly localized distribution in real space. Let us assume a target resolution of some δr ≈ 0,1 ˚ A in a cubic lattice of a ≈ 5 ˚ A. This corresponds in k-space to δk = 2π/δr ≈ 63 ˚ A−1 and 2πa = 2π/a ≈ 1,3 ˚ A−1. Thus, in the sum over reciprocal lattice vectors, G = 2πa ˜ n, we need n ∈ [−50, +50] or about 106 planewaves. Solving the secular equations means obtaining the lowest eigenvalues and eigenvectors of a 106 × 106 matrix plus iterating to self-consistency, and that work has to be repeated on each q-point. There are two main ways, however, to turn practical the use of planewaves. The first method involves the use of pseudopotentials that simulate the effect of the core electrons, thus limiting the crystal calculation to the valence electrons. This reduces drastically the spacial resolution and the number of planewaves required. A different strategy consist in modifying the basis set, adding core tailored components to the planewaves like in the OPW (Orthogonalized PWs) and APW (Augmented PWs) methods, or substituting them with exponential (STO, Slater Type functions) or gaussian functions (GTO, Gaussian Type functions) like in the LCAO (Linear Combination of Atomic like functions) method. Treatment of the lattice potential. Atomic form factors. A great ecomomy is achieved in the treatment of the potential terms if they can be expressed as a sum of atomic c V. Lua˜ na, QTC Murcia 2008 (173)
waves contributions: V (r) = L i Vi(r − Ri − L) (204) where L sums over unit cells, i sums over the atoms in one unit cell, and Ri is the nuclear position of i relative to the origin of its unit cell. The Fourier coefficients for this potential are Vg = V V (r)e−igrdr = L i V Vi(r − Ri − L)e−igrdr = r = r − Ri − L; dr = dr ; r ∈ V ; r ∈ R3. = L e−igL =1 i e−igRi R3 Vi(r )e−igr dr = Ncell i vi(g)e−igRi (205) where we have taken into account e−igL = 1 because g and L belong in the k-lattice and r-lattice, respectively. Equation 205 makes clear that Vg is an extensive quantity, proportional to the number of unit cells in the crystal. On most cases we really want the intensive Vg per cell. The vi(g) function is called the atomic form factor, whereas e−igr is the geometric term, as it only depends on the position of the nuclei within the cell. If Vi(r ) = Vi(|r |) then vi(g) = vi(|g|) and the computational effort is accordingly reduced. c V. Lua˜ na, QTC Murcia 2008 (174)
Waves (OPW) OPWs, introduced by Herring in 1940 [51], were the basis for the first quantitative calculations, in the 40s and early 50s, of bands in general materials. Today OPWs have been surpassed by other methods, but the technique is still regarded as the natural introduction to pseudopotentials in solid state physics. As we have discussed before, plane waves are inefficient to represent the well localized electron density around nuclei. Not only the core orbitals, but even the valence wavefunctions require very high k components due to their internal nodal structure caused by the required orthogonality to the core levels. The idea behind OPWs is fulfilling the valence-to-core orthogonality from the beginning, so that the basis functions do only need to expand the valence density out of the near-core regions. Let |q be a planewave and |ψc one of the core states that we want to remove from the solution space of the crystal orbital Schr¨ odinger equation. The OPW associated to |q is |OPW, q = |q − c |ψc ψc|q . (206) If the |ψc were a complete set of states c |ψc ψc| = ˆ 1 and the OPW would vanish. This is clearly false in our case, but we expect that the core states show a strong localization around the nuclear positions and that OPWs remain small there. Working with OPWs and the normal one particle hamiltonian is equivalent to working with PWs c V. Lua˜ na, QTC Murcia 2008 (175)
modified hamiltonian: (ˆ h − q) |OPW, q = − 2 2m ∇2 r + ˆ V (r) − q |q − c |ψc ψc|q = − 2 2m ∇2 r |q + ( ˆ V − q) |q − c ψc|q ≈ ( c − q) |ψc (ˆ h − q) |ψc = − 2 2m ∇2 r + ˆ V − c ( c − q) |ψc ψc| − q |q = (ˆ hps − q) |q (207) where ˆ hps = − 2 2m ∇2 r + ˆ Vps; ˆ Vps = ˆ V − c ( c − q) |ψc ψc| . (208) In words, the lattice potential ˆ V (r) is replaced by a lattice pseudopotential that contains the orthogonality condition of the PWs to the core sates. This pseudopotential tends to be smoother on the core regions that the original lattice potential, thus allowing for a significant reduction in the number of PWs required. On the other hand, ˆ Vps is a non-local operator that, in addition, depends on the unknown eigenvalue of the crystal state upon which it is applied. This introduces a further complication on the one particle equation, but this is more than compensated by the reduction in the PW basis set. c V. Lua˜ na, QTC Murcia 2008 (176)
initio pseudopotentials have a very long tradition in many areas of atomic, molecular, nuclear and condensed matter quantum problems. Chemists will stress its origin on the core-valence and sigma-pi separability problems, and the names of Lykos, Parr, McWeeny and Huzinaga, fathers of the Electron Separability Theory, are unavoidable. Solid state physicists will prefer pointing to the very influential 1959 article by Phillips and Kleinman and quickly step to the modern soft and ultrasoft potentials by Vanderbilt, Hamann et al., or Troullier-Martins schools. Common to all the traditions and schools, core pseudopotentials are built to reproduce the valence levels of a reference all electron atomic calculation. Let { AE nlm , ψAE nlm (r)} be the reference orbitals, solution to the atomic one electron equation − 2 2m ∇2 r + V (r) ψAE nlm (r) = AE nlm ψAE nlm (r). (209) The pseudopotential equation − 2 2m ∇2 r + V ps l ˜ ψps nlm (r) = ps nlm ˜ ψps nlm (r) (210) differs from the all electron counterpart in several important aspects: 1. Core states are projected out from the solution space of eq. 210. Valence states are kept untouched, i.e. ps = AE. c V. Lua˜ na, QTC Murcia 2008 (177)
of the pseudofunction ψps is modified by removing out the inner nodal structure of the true valence function. ψps is kept identical to ψAE beyond a given cutoff radius Rc, but the inner wiggles are changed into a smooth, nodeless, function for r < Rc. 3. In the norm-conserving pseudopotentials ψps and ψAE integrate identically for r < Rc. This condition is removed for the soft and ultrasoft potentials. Continuity conditions must be applied at Rc. c V. Lua˜ na, QTC Murcia 2008 (178)
on l. This can be achieved in several ways: Local ps: ˆ Vps = U(r) (local in r). Semilocal ps: ˆ Vps = l Ul(r) ˆ Pl (local in r, nonlocal in θ, φ). Nonlocal separable ps: ˆ Vps = UL+1(r) + L l=0 |Ylm Ul(r) Ylm|. General ps: ˆ Vps = UL+1(r) + t,t L l=0 Dtt l |βtlm βt lm |. 5. Pseudopotentials provide a cheap way to introduce relativistic effects. Obtaining a good pseudopotential is a delicated handicraft that involves: 1. Deciding which atomic orbitals are kept in the valence. For instance, 3spd4s are typically the valence in the first transition metal group. 2. Creating the pseudofunctions (cutoff radius, smooth inner zone, continuity conditions, norm conserving, . . . ). 3. Determining the pseudopotentials by inverting eq. 210. 4. Checking for the occurrence of anomalies: e.g. ghost states. 5. Analyzing the transferability of the potentials for electronic states of the atom different from the states used as reference. This is a good test for transferability to molecules and crystals. 6. Testing the potentials in the solid. c V. Lua˜ na, QTC Murcia 2008 (179)
sure that the pseudopotential scattering properties remain correct for nearby nl (Hamann et al., 1979 [52]). Norm-conserving potentials are accepted by most solid state codes. Soft (Troullier-Martins [53] are currently the most popular) and ultrasoft (i.e. Vanderbilt [54]) have a clear computational advantage by requiring a reduced number of PW’s. In other words, the energy cutoff for the PW included in the solid state calculation can be considerably reduced. Some extra coding is required in the solid-state program, however, and not all packages accept ultrasoft potentials. There is a close relationship between the ultrasoft potentials and the Projector Augmented-Wave method [55]. In fact, the PAW technique provides a way for coding the ultrasoft potentials [56]. Some web resources: • Martins site: bohr.inesc-mn.pt/~jlm/. • Vanderbilt site: www.physics.rutgers.edu/~dhv/. • The Octopus code: www.tddft.org/programs/octopus/wiki/index.php/Main_Page. • A nice Vanderbilt’s report on pseudopotentials: www.physics.rutgers.edu/~dhv/talks/ bangalore-july06.pdf. c V. Lua˜ na, QTC Murcia 2008 (180)
solid state codes Abinit: GPL, www.abinit.org, X. Gonze et al. (Universit´ e Catholique de Louvain; Corning Inc.; etc). Pwscf: GPL, www.pwscf.org, S. Baroni et al. (Trieste, . . . ). Wien2k: distributed as source for a small subscription, www.wien2k.at, P. Blaha (K. Schwartz) et al. (Technical University Wien, . . . ) Siesta (Spanish Initiative for Electronic Simulations with Thousands of Atoms): distributed as source for a small subscription, www.uam.es/siesta/, E. Artacho (U. Cambridge), P. Ordej´ on (ICM, Barcelona), J. Junquera (U. Cantabria), D. S´ anchez Portal (UPV), J. M. Soler (UAM), . . . et al. crystal06: only binary is distributed (06) or source could not be modified (03 and 98 versions), www.crystal.unito.it, R. Dovesi (U. Torino), V.R. Saunders (Daresbury Lab., U. Cambridge) et al. References Kantorovich, 2004 [33]; Kaxiras, 2003 [34]; Kittel, 2005 [35]; Marder, 2000 [36]; Martin, 2004 [37]; McQuarrie, 1976 [38]; Pueyo, 2005 [39]; c V. Lua˜ na, QTC Murcia 2008 (182)
Mermin, Solid State Physics (Saunders College, Philadelphia, 1976), pp. 826+xxi. [2] H. K. D. H. Bhadeshia, Worked examples in the Geometry of Crystals, 2nd ed. (The Institute of Materials, London, UK, 2006), pp. 104+iv. [3] G. Burns and A. M. Glazer, Space groups for solid state scientists, 2nd ed. (Academic Press, San Diego, CA, 1990), pp. 343+vii. [4] C. Giacovazzo, H. L. Monaco, G. Artioli, D. Viterbo, G. Ferraris, G. Gilli, G. Zanotti, and M. Catti, Fundamentals of Crystallography, Vol. 2 of IUCr texts on crystallography, 2nd ed. (Oxford UP, Oxford, UK, 2002), pp. 825+xx. [5] International Tables for X-Ray Crystallography. A. Space-group symmetry, Vol. A of International Tables for X-Ray Crystallography, edited by T. Hanh (D. Reidel, Dordrecht, Holland, 1983), p. 894. [6] C. J. Bradley and A. P. Cracknell, The mathematical theory of symmetry in solids (Clarendon, Oxford, UK, 1972), pp. 745+xii. [7] S. Bhagavantam, Crystal symmetry and physical properties (Academic, New York, 1966), p. 227. [8] J. F. Nye, Physical Properties of Crystals (Oxford UP, Oxford, UK, 1985), p. xxx, republication of the 1957 classic. [9] M. Catti, Acta Cryst. A 41, 494 (1985). [10] M. Catti, Acta Cryst. A 45, 20 (1989). [11] M. Catti, R. Dovesi, A. Pavese, and V. R. Saunders, J. Phys.: Condens. Matter 3, 4151 (1991). [12] F. Jona and P. M. Marcus, Phys. Rev. B 63, 094113 (2001). [13] V. Lua˜ na, J. M. Recio, and L. Pueyo, Phys. Rev. B 42, 1791 (1990). [14] M. A. Blanco, E. Francisco, and V. Lua˜ na, Comput. Phys. Commun. 158, 57 (2004), source code distributed by the CPC program library: http://cpc.cs.qub.ac.uk/summaries/ADSY. [15] M. A. Blanco, V. Lua˜ na, and A. M. Pend´ as, Comput. Phys. Commun. 103, 287 (1997). [16] J. D. Gale and A. L. Rohl, Molecular Simulation 29, 291 (2003). [17] V. Lua˜ na and L. Pueyo, Phys. Rev. B 41, 3800 (1990). c V. Lua˜ na, QTC Murcia 2008 (184)
J. Chem. Phys. 97, 6544 (1992). [19] V. Lua˜ na, M. Fl´ orez, and L. Pueyo, J. Chem. Phys. 99, 7970 (1993). [20] A. M. Pend´ as, V. Lua˜ na, J. M. Recio, M. Fl´ orez, E. Francisco, M. A. Blanco, and L. N. Kantorovich, Phys. Rev. B 49, 3066 (1994). [21] M. ´ Alvarez Blanco, Tesis doctoral, Universidad de Oviedo, 1997, director: V´ ıctor Lua˜ na Cabal. [22] T. Schlick, Molecular Modeling and Simulation (Springer Verlag, New York, 2002), pp. 634+xliv. [23] R. C. Mota, P. S. Bran´ ıcio, and J. P. Rino, Europhys. Lett. 76, 836 (2006). [24] M. S. Daw and M. I. Baskes, Phys. Rev. Lett. 50, 1285 (1983). [25] A. P. Sutton and J. Chen, Philos. Mag. Lett. 61, 139 (1990). [26] F. Calvo, J. P. K. Doye, and P. J. Wales, J. Chem. Phys. 114, 7312 (2001). [27] J. P. K. Doye and F. Calvo, J. Chem. Phys. 116, 8307 (2002). [28] E. G. Noya and J. P. K. Doye, J. Chem. Phys. 124, 104503 (2006). [29] F. H. Stillinger, J. Chem. Phys. 115, 5208 (2001). [30] S. Somasi, B. Khomami, and R. Lovett, J. Chem. Phys. 113, 4320 (2000). [31] P. Krishna and D. Pandey, Close-Packed Structures (University College Cardiff Press for the IUCr, Cardiff, Wales, 1981), p. 24. [32] A. Damascelli, Z. Hussain, and Z.-X. Shen, Rev. Mod. Phys. 75, 473 (2003), free version on http://arxiv.org/abs/cond-mat/0208504. [33] L. Kantorovich, Quantum Theory of the Solid State: An Introduction (Kluwer, Dordrecht, The Netherlands, 2004), pp. 626+xxv. [34] E. Kaxiras, Atomic and Electronic Structure of Solids (Cambridge University Press, Cambridge, UK, 2003), pp. 676+xx. [35] C. Kittel, Solid State Physics, 8th ed. (Wiley, New York, 2005), p. 705. [36] M. P. Marder, Condensed Matter Physics (Wiley-Interscience, New York, 2000), pp. 895+xxvi. [37] R. M. Martin, Electronic Structure: Basic theory and practical methods (Cambridge, Cambridge, UK, 2004), pp. 624+xxiii. [38] D. A. McQuarrie, Statistical Mechanics (Harper & Row, New York, 1976), pp. 641+xv. c V. Lua˜ na, QTC Murcia 2008 (185)
superficies y s´ olidos. I. Estructura electr´ onica de los s´ olidos, http://web.uniovi.es/qcg/d-EstrElSol/, 2005. [40] J. C. Slater and G. F. Koster, Phys. Rev. 94, 1498 (1954). [41] C. M. Goringe, D. R. Bowler, and E. Hern´ andez, Rep. Prog. Phys. 60, 1447 (1997). [42] W. A. Harrison, Electronic structure and the properties of solids, dover republication of the 1980 edition published by w. h. freeman, san francisco ed. (Dover, New York, 1989), pp. 582+xv. [43] W. A. Harrison, Elementary electronic structure, revised edition ed. (World Scientific, New Jersey, 2005), pp. 836+xx. [44] P. Y. Yu and M. Cardona, Fundamentals of semiconductors, 3rd ed. (Springer, Berlin, Germany, 2001), pp. 639+xviii. [45] P. Vogl, H. J. Hjalmarson, and J. D. Dow, J. Phys. Chem. Solids 44, 365 (1983). [46] L. van Hove, Phys. Rev. 89, 1189 (1953). [47] H. J. Monkhorst and J. D. Pack, Phys. Rev. B 13, 5188 (1976). [48] M. Methfessel and A. T. Paxton, Phys. Rev. B 40, 3616 (1989). [49] J. Moreno and J. M. Soler, Phys. Rev. B 45, 13891 (1992). [50] A. Otero de la Roza, Tesis de Licenciatura, Universidad de Oviedo, 2007, director: V´ ıctor Lua˜ na. [51] W. C. Herring, Phys. Rev. 57, 1169 (1940). [52] D. R. Hamann, M. Schl¨ uter, and C. Chiang, Phys. Rev. Lett. 43, 1494 (1979). [53] N. Troullier and J. L. Martins, Phys. Rev. B 43, 1993 (1991). [54] D. Vanderbilt, Phys. Rev. B 41, 7892 (1990). [55] P. E. Bl¨ ochl, Phys. Rev. B 50, 17953 (1994). [56] G. Kresse and D. Joubert, Phys. Rev. B 59, 1758 (1999). c V. Lua˜ na, QTC Murcia 2008 (186)
{kind=link}
{kind=link}
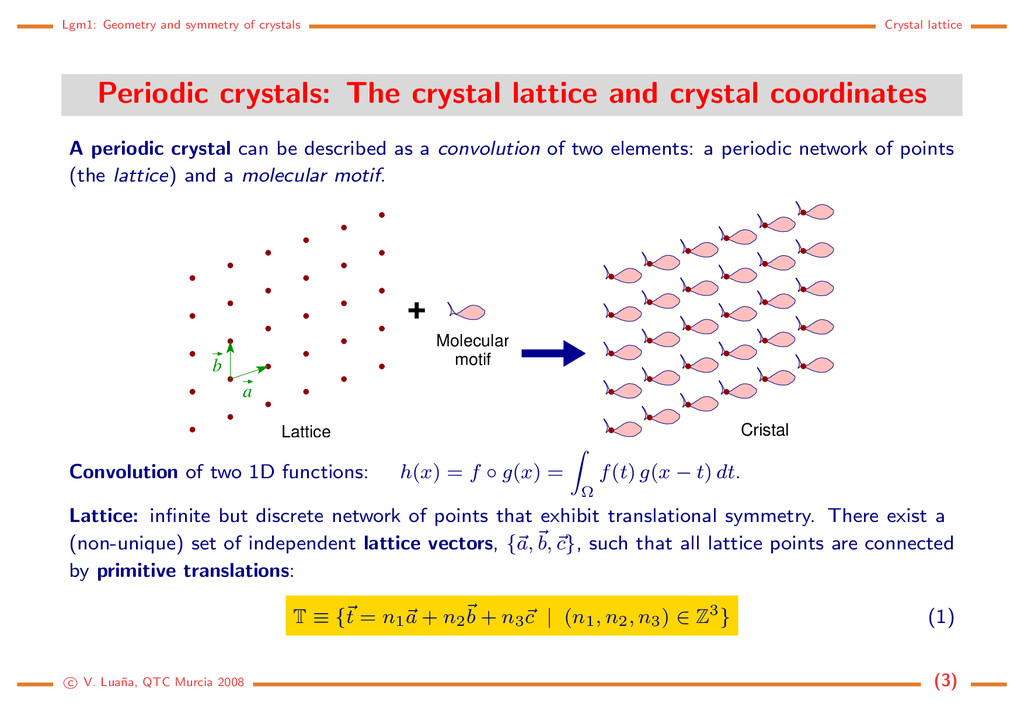{kind=link}
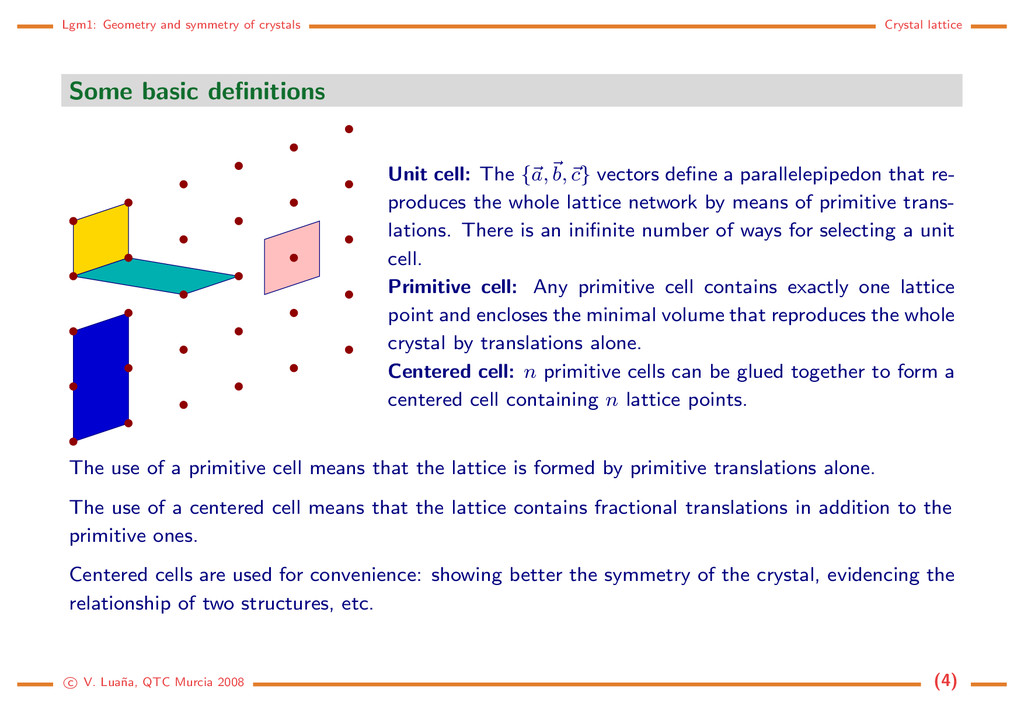{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
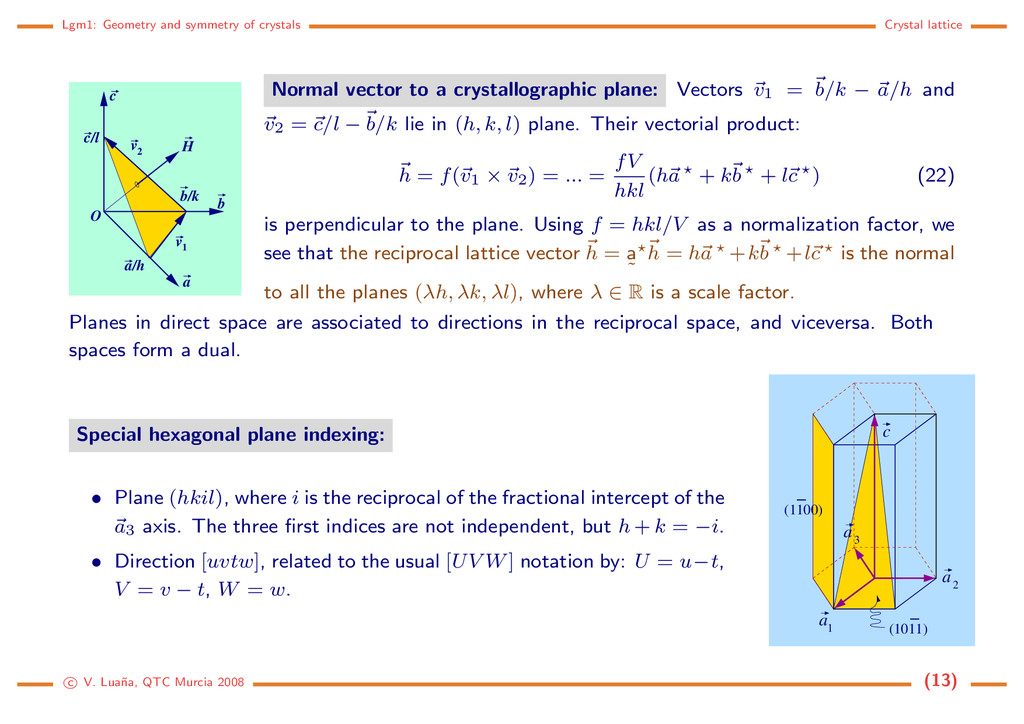{kind=link}
{kind=link}
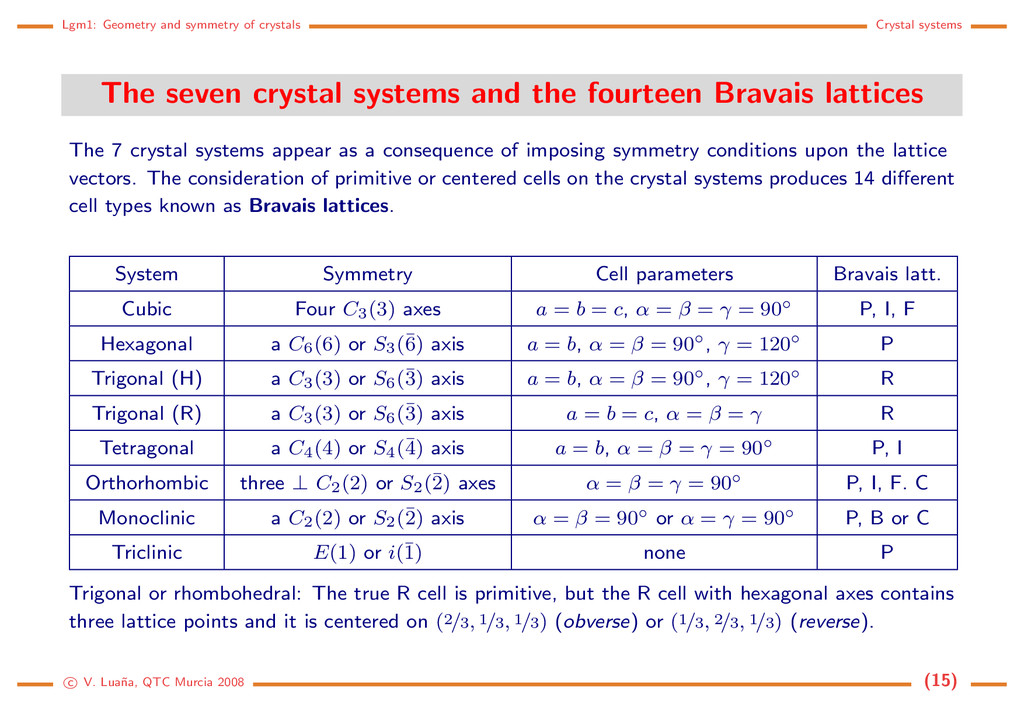{kind=link}
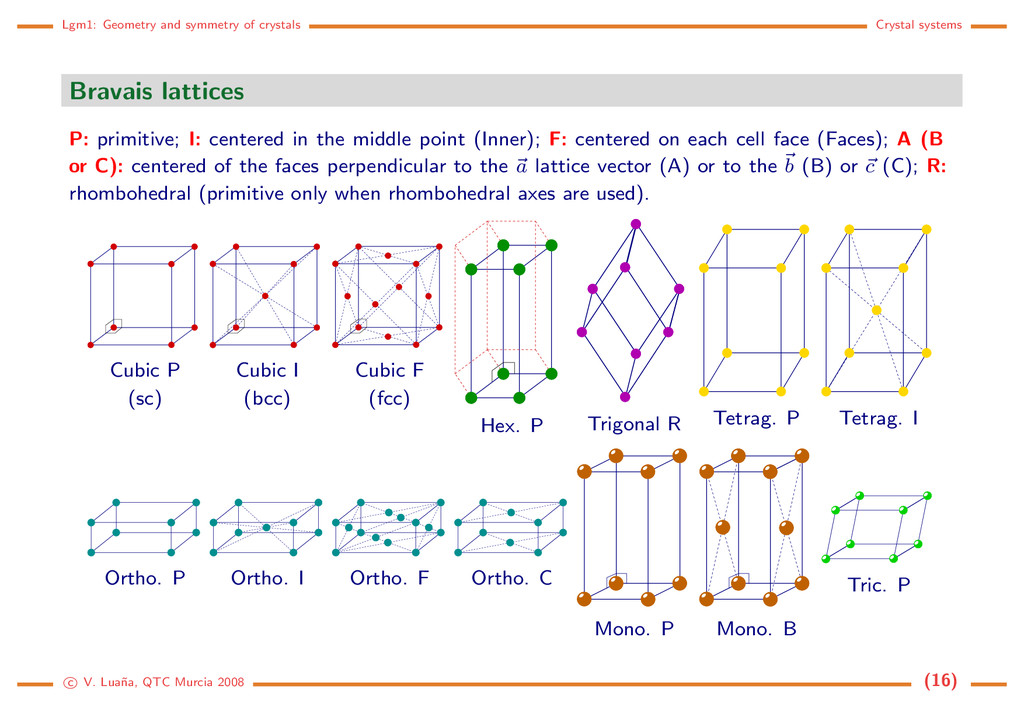{kind=link}
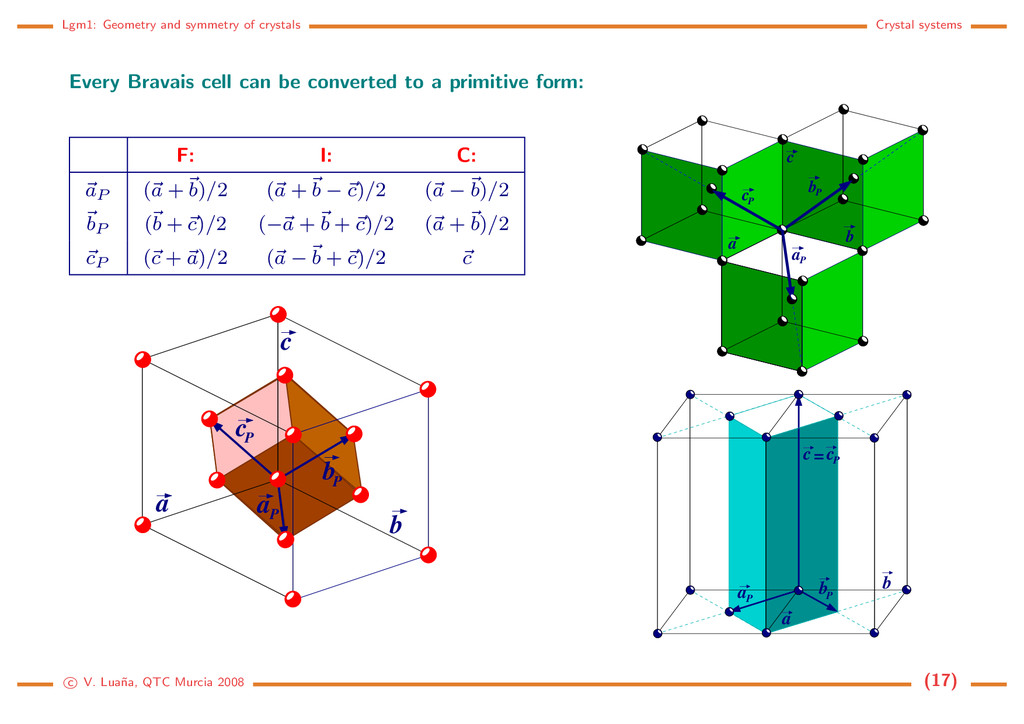{kind=link}
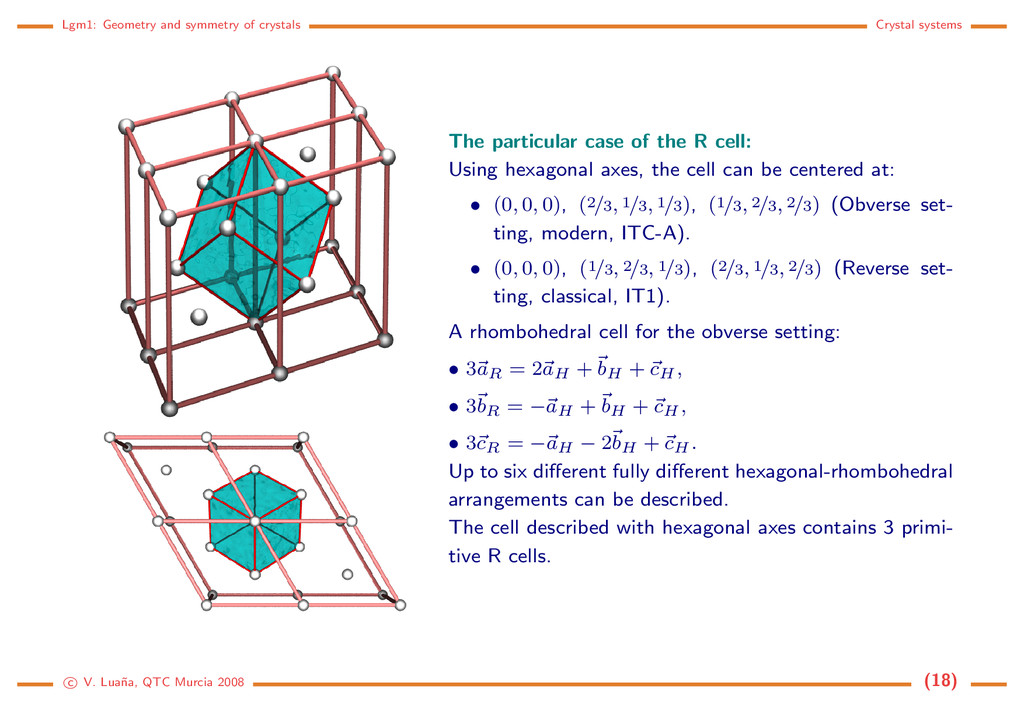{kind=link}
{kind=link}
{kind=link}
{kind=link}
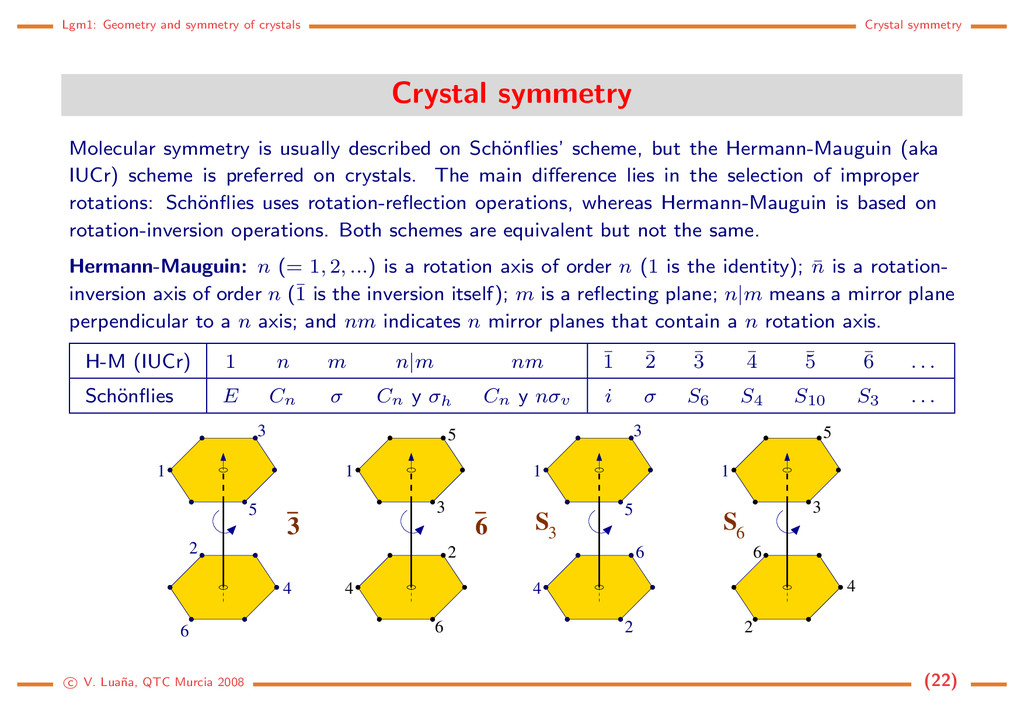{kind=link}
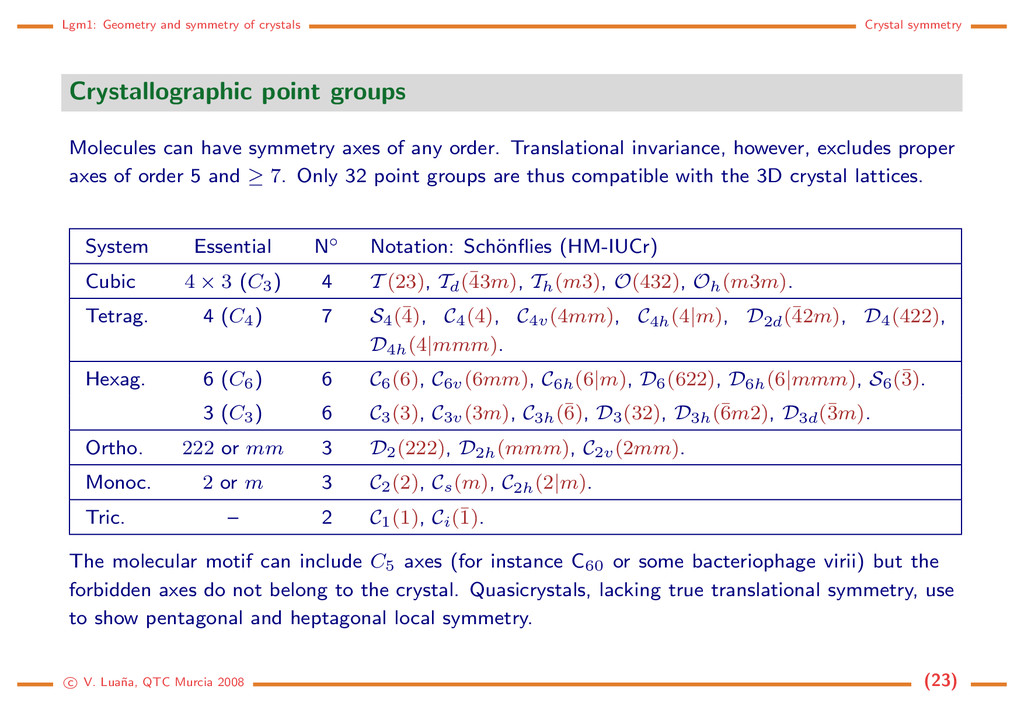{kind=link}
{kind=link}
{kind=link}
{kind=link}
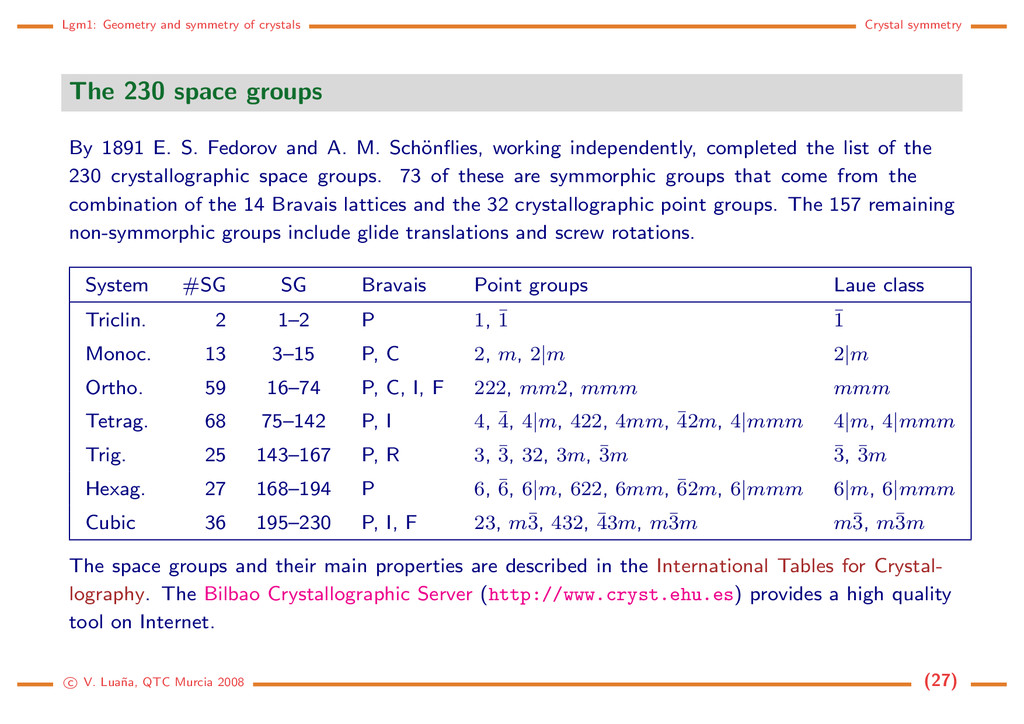{kind=link}
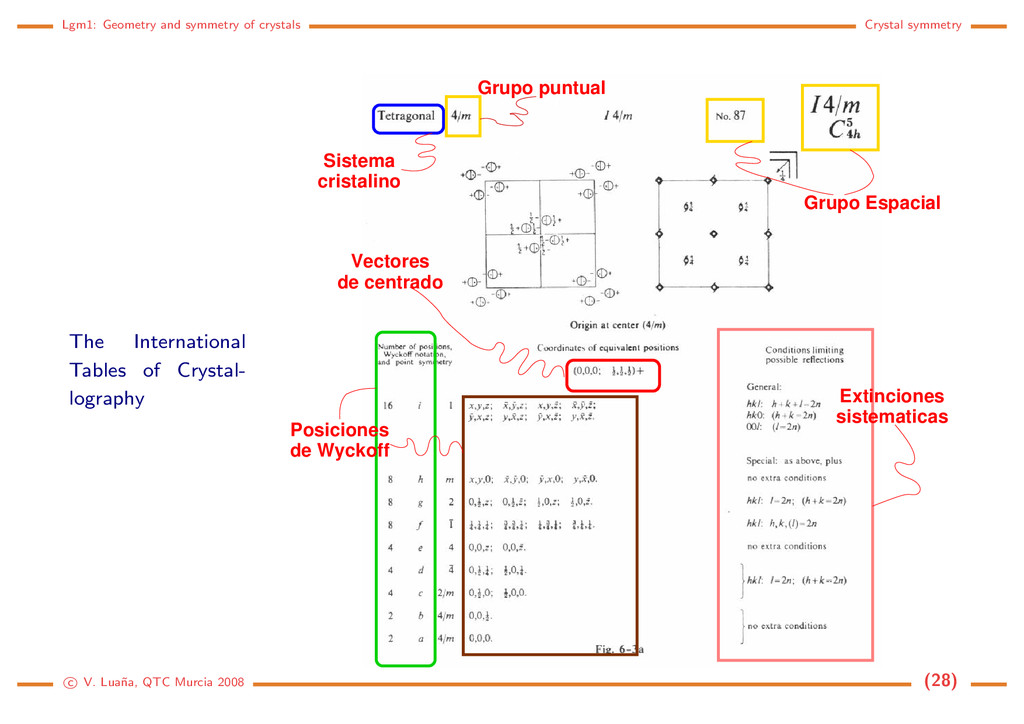{kind=link}
{kind=link}
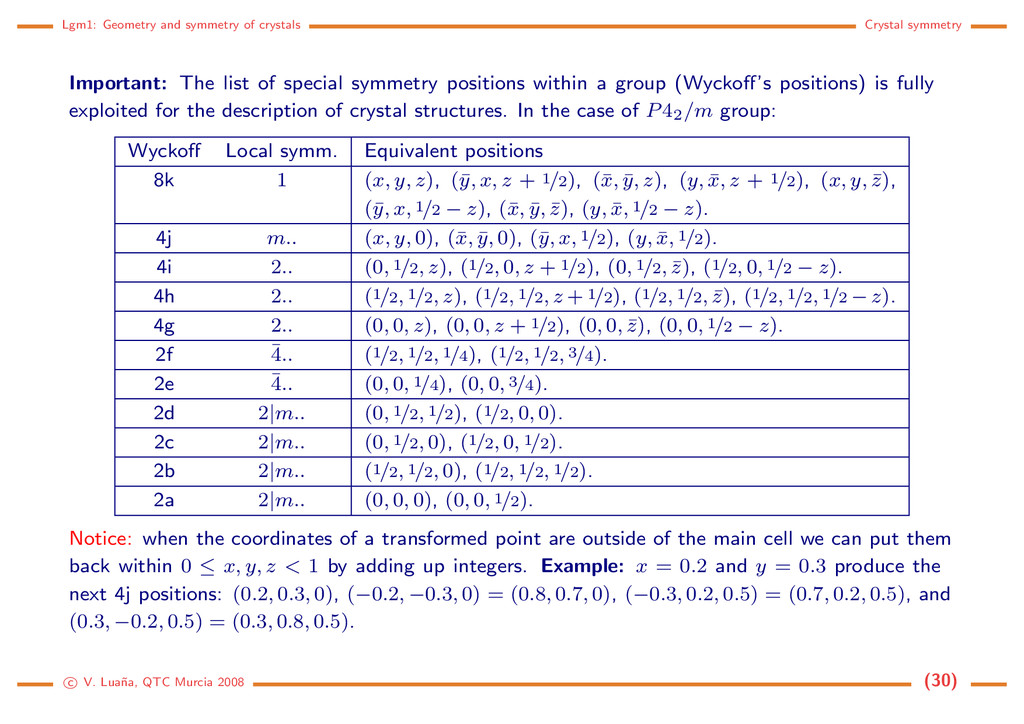{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
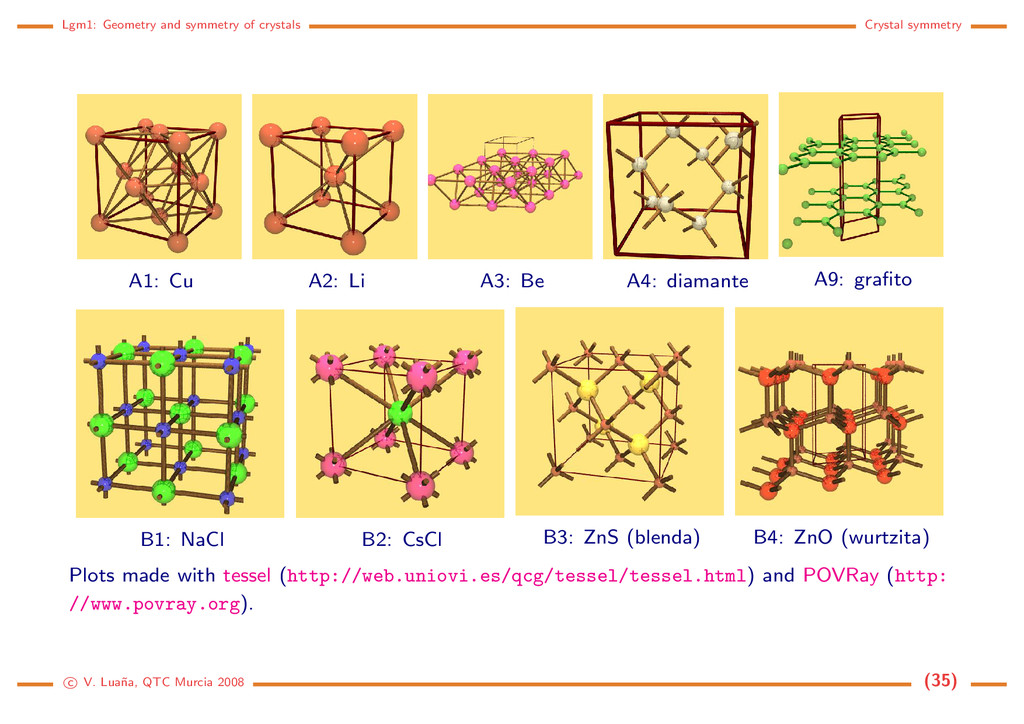{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
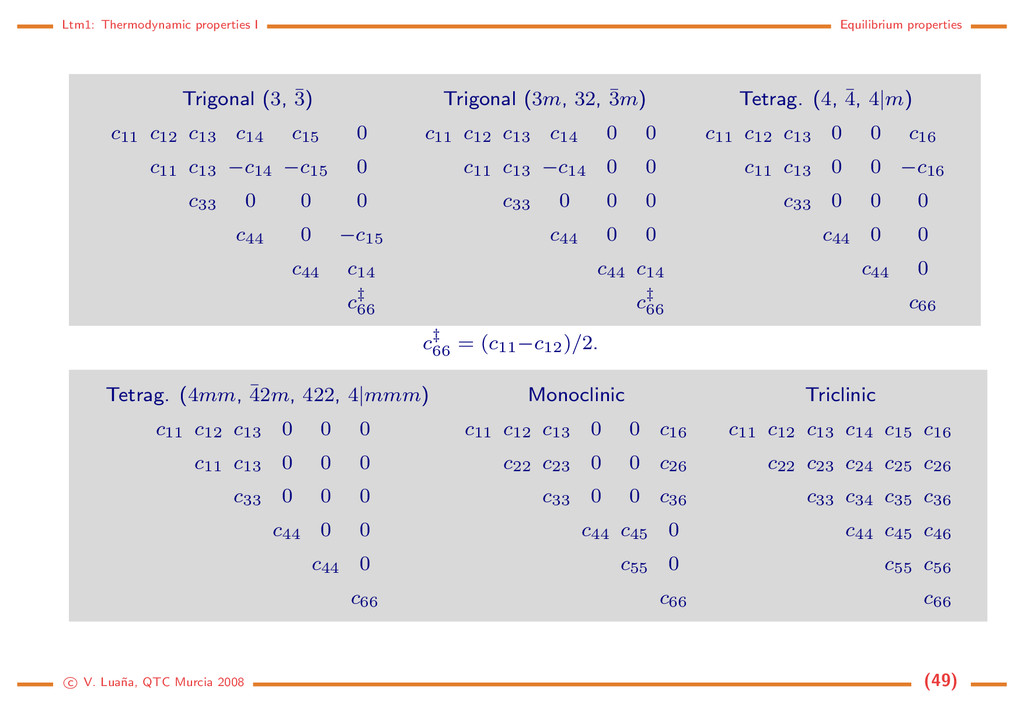{kind=link}
{kind=link}
![Ltm1: Thermodynamic properties I Equilibrium properties Example [11]: Cubic CaF2](https://files.speakerdeck.com/presentations/e45d9cf068ec013133b97691c1dda2be/slide_50.jpg){kind=link}
{kind=link}
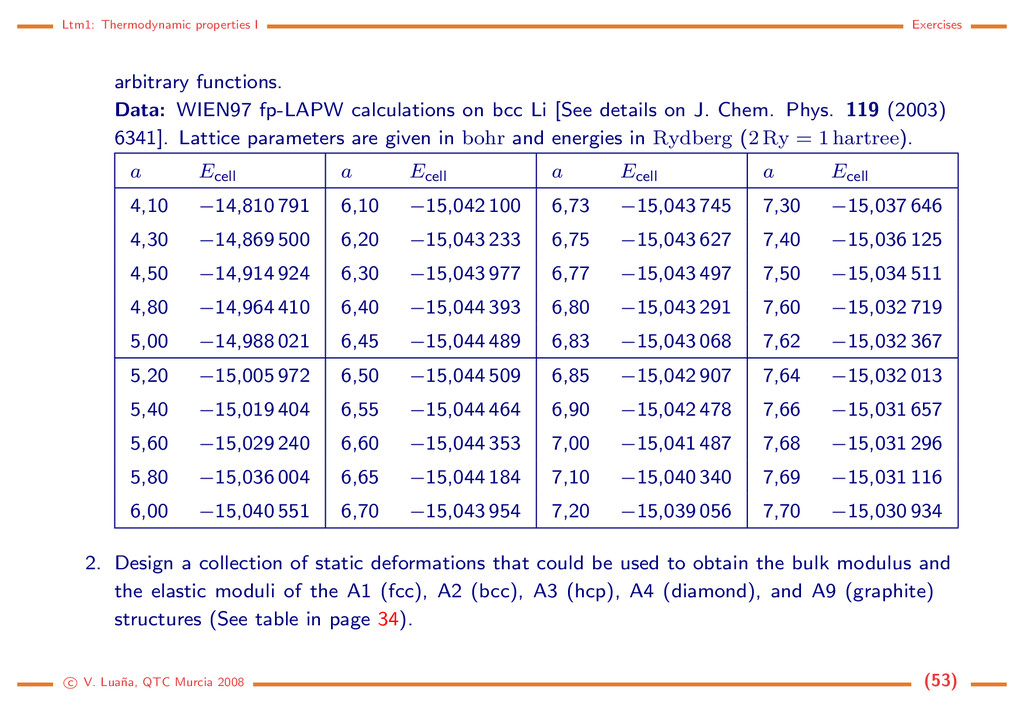{kind=link}
{kind=link}
{kind=link}
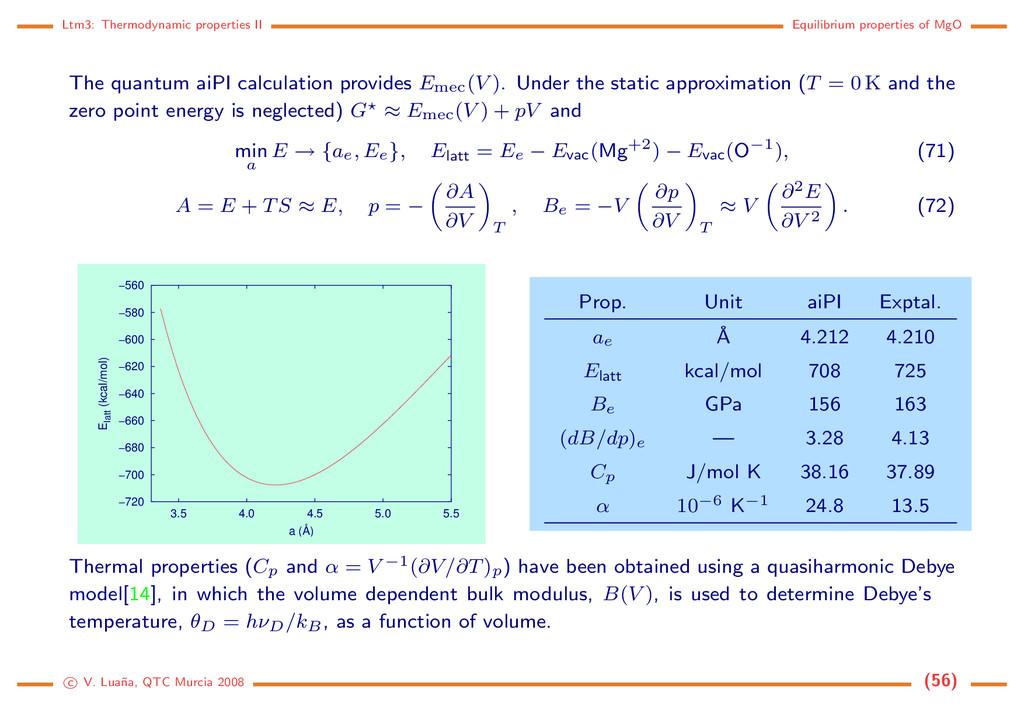{kind=link}
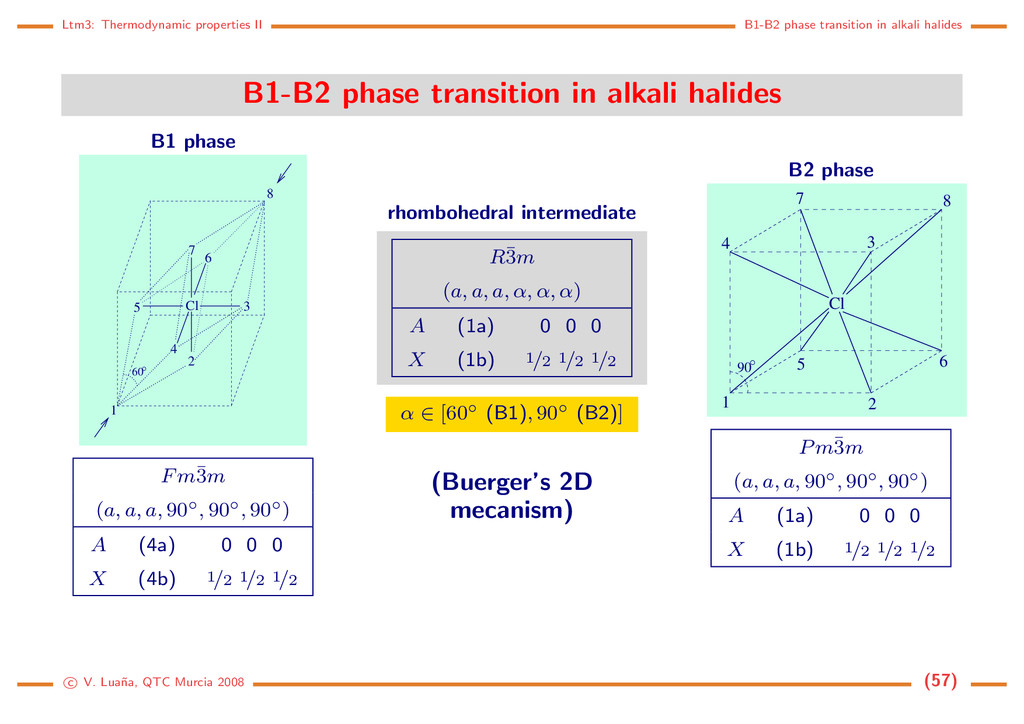{kind=link}
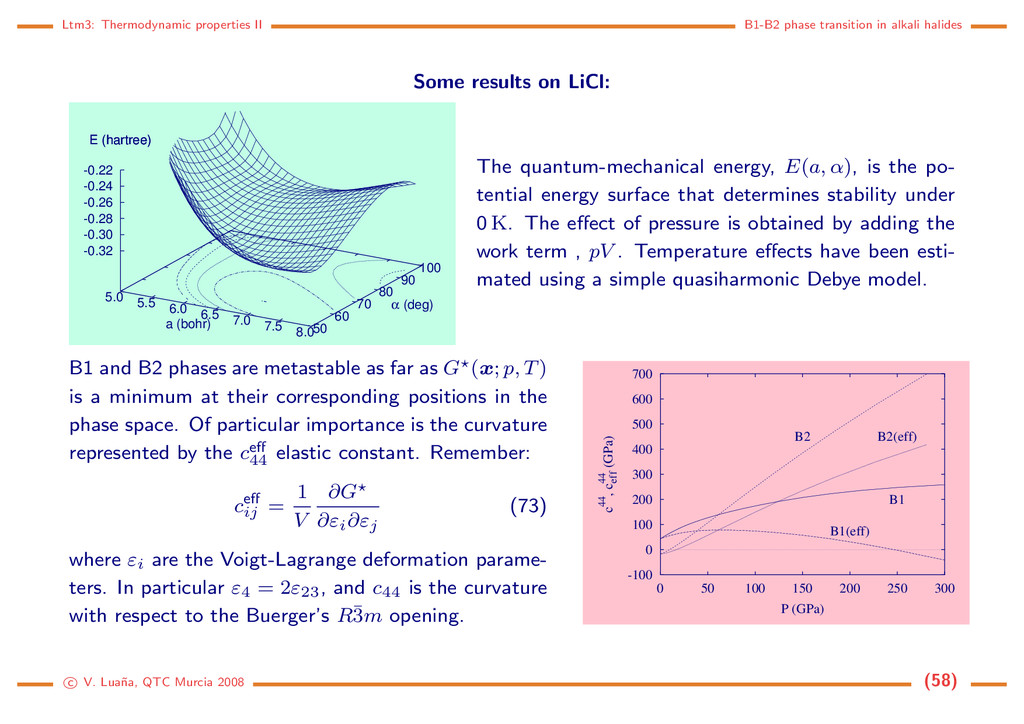{kind=link}
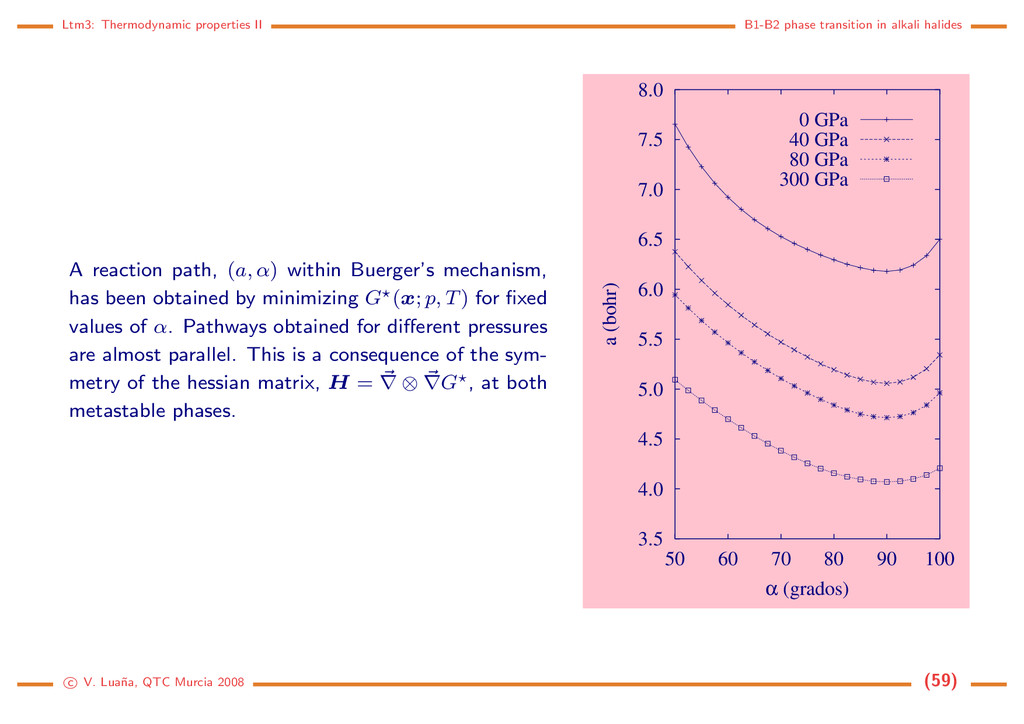{kind=link}
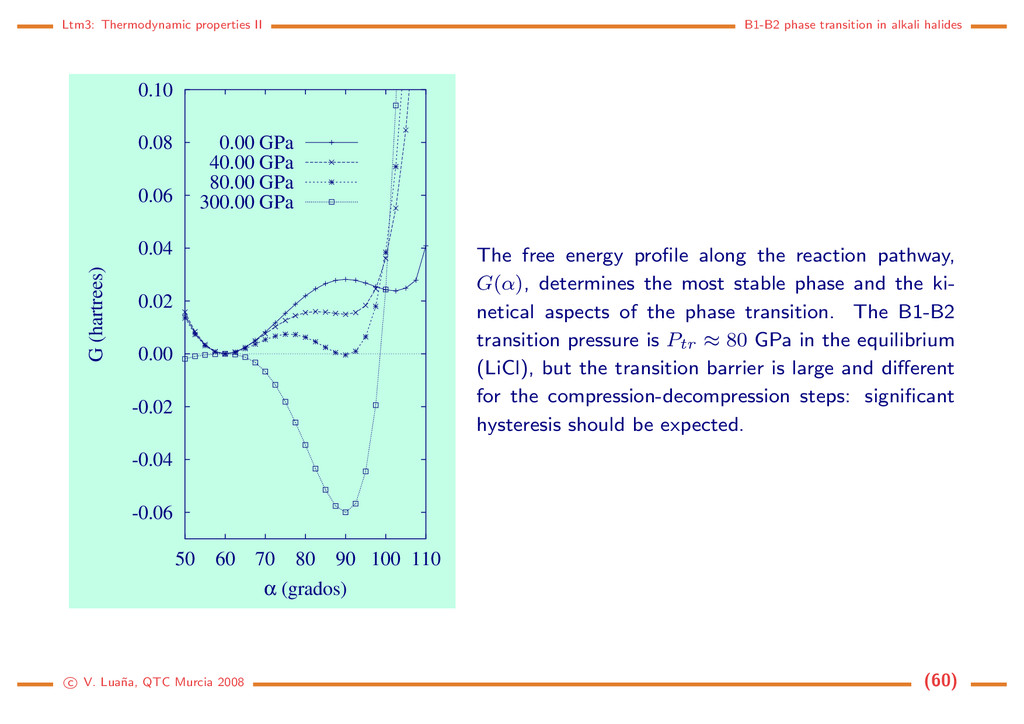{kind=link}
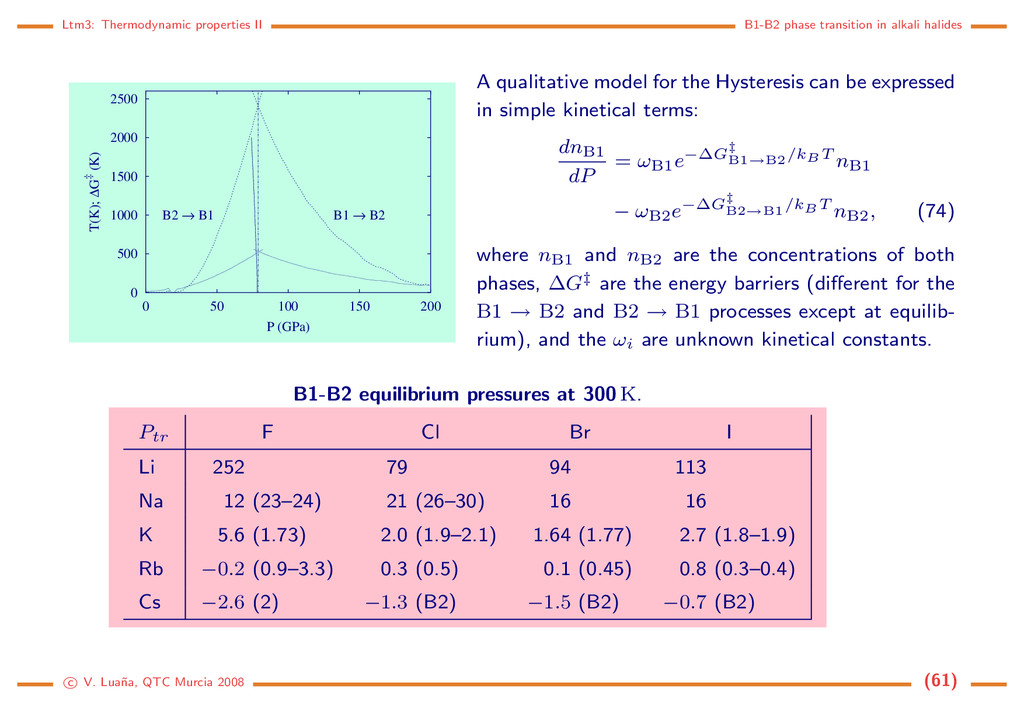{kind=link}
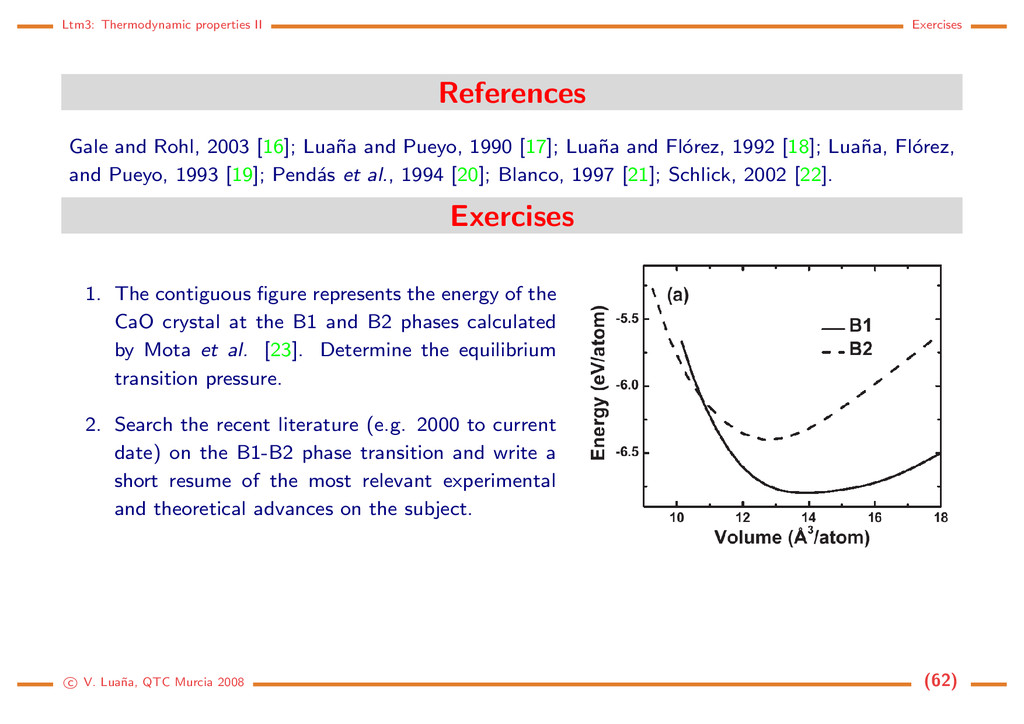{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
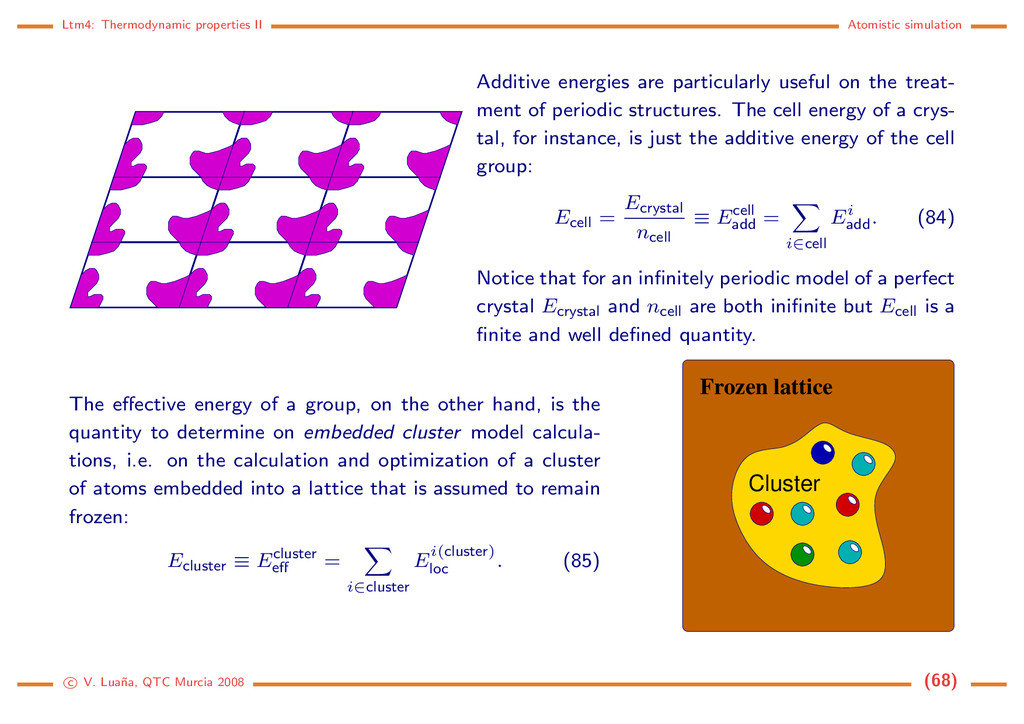{kind=link}
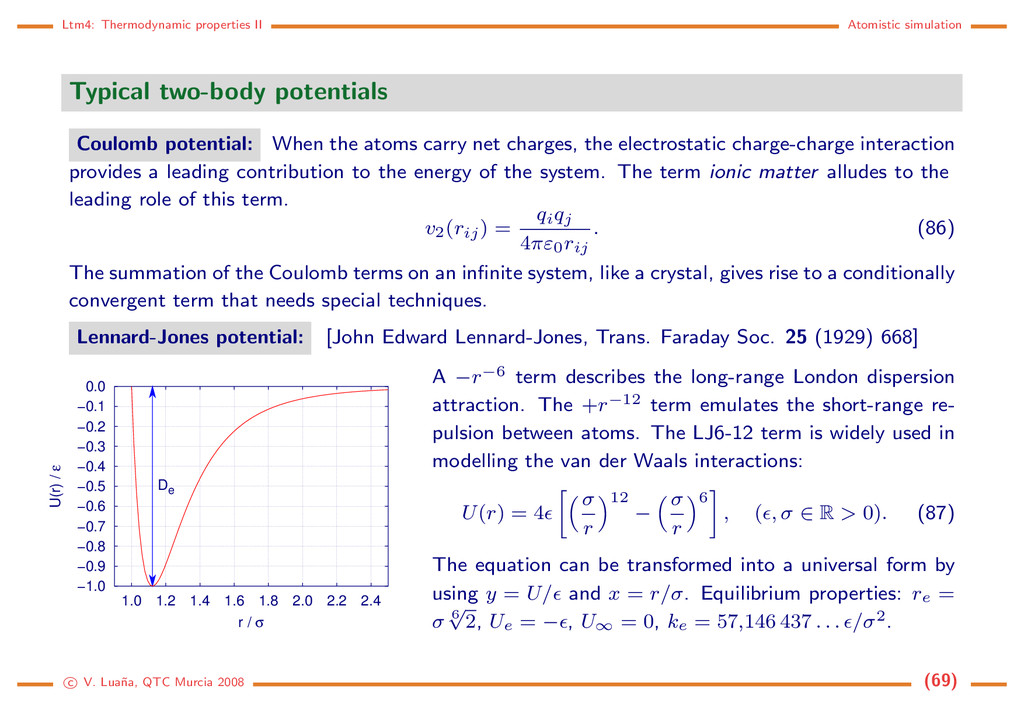{kind=link}
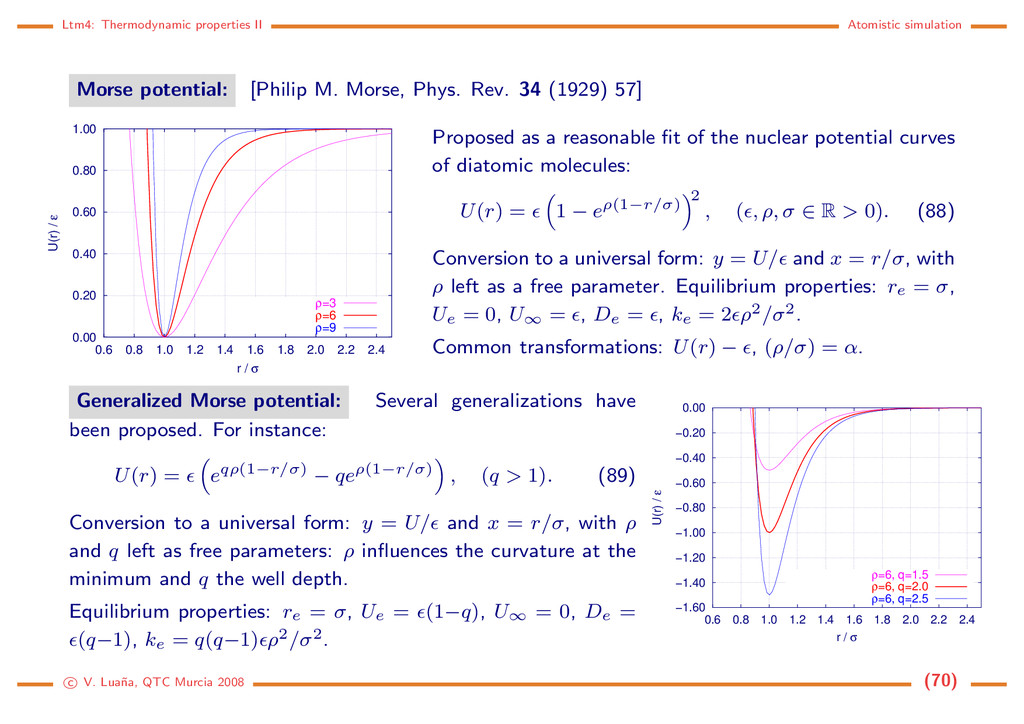{kind=link}
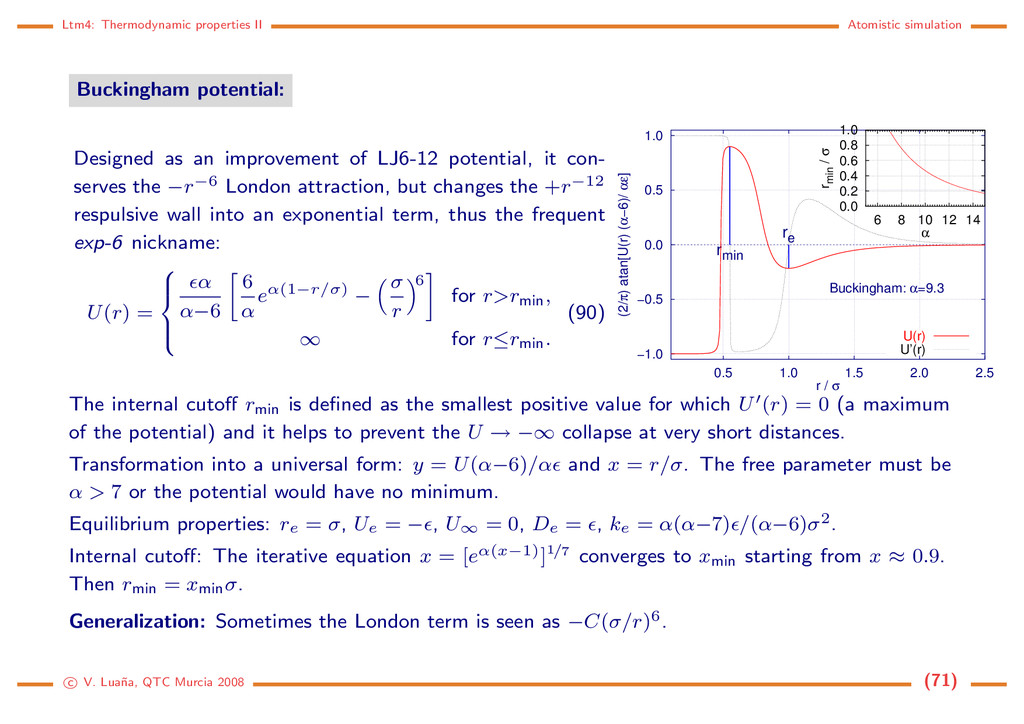{kind=link}
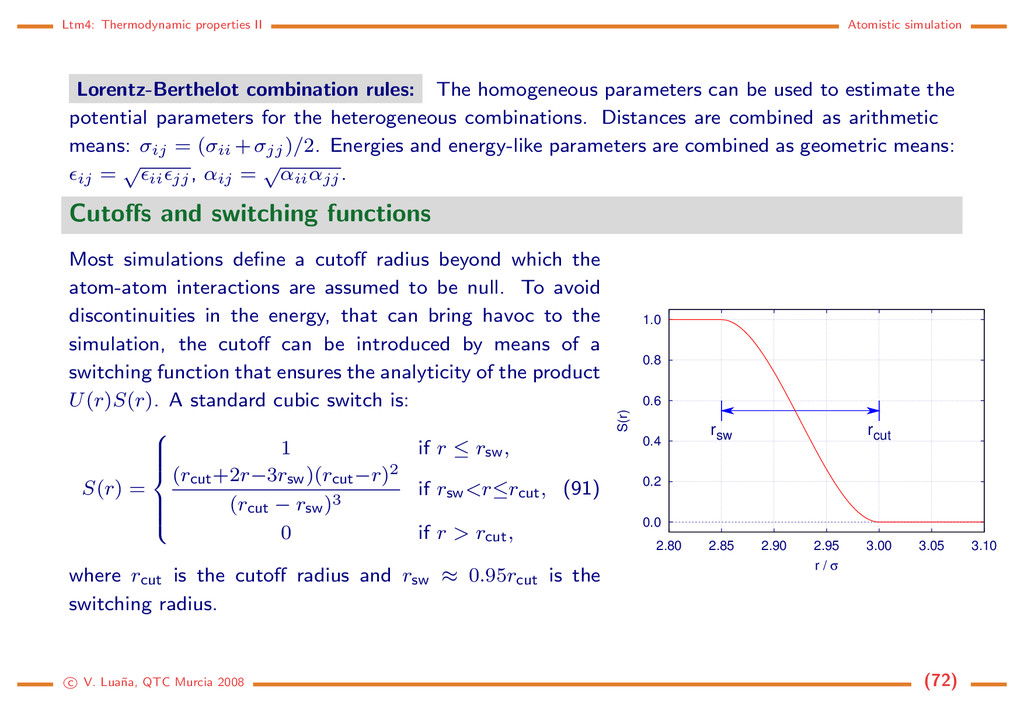{kind=link}
{kind=link}
{kind=link}
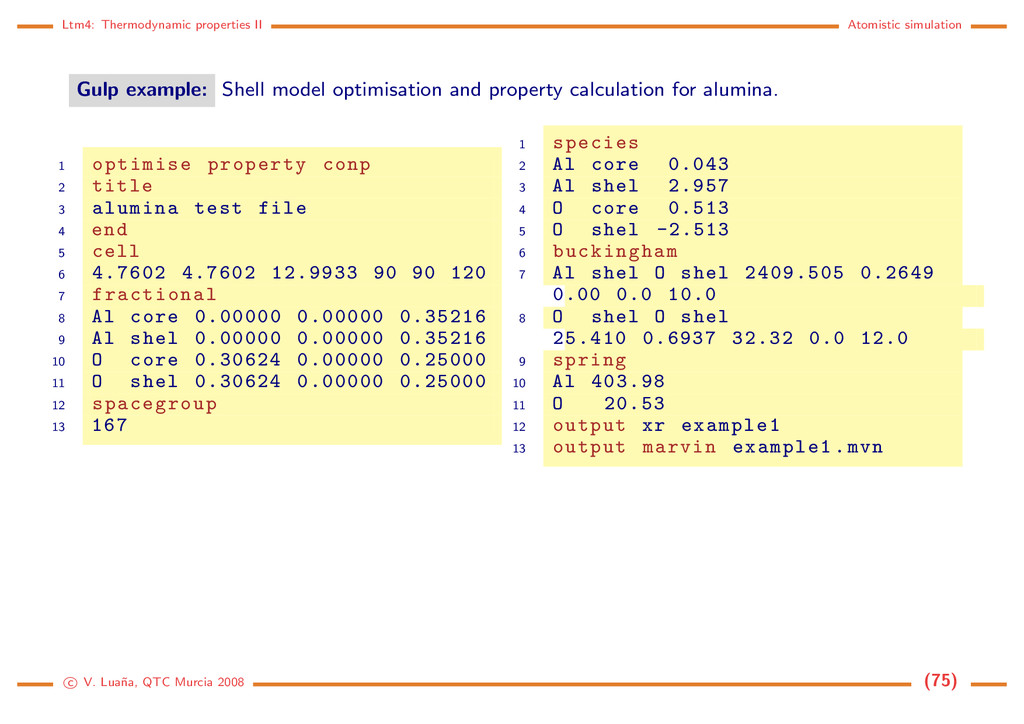{kind=link}
{kind=link}
{kind=link}
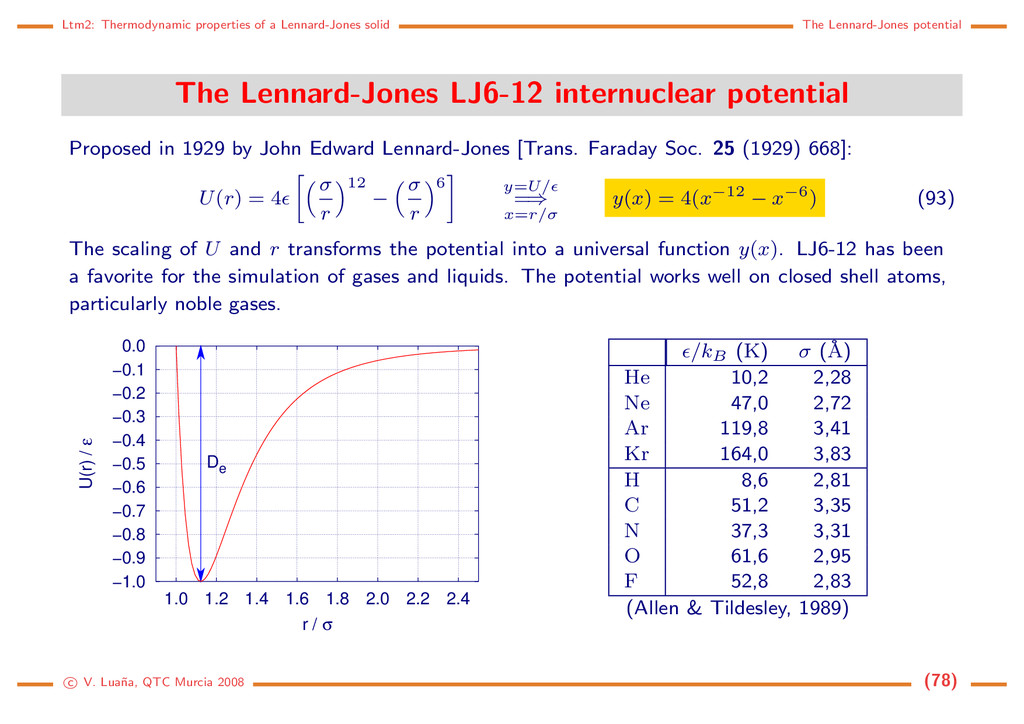{kind=link}
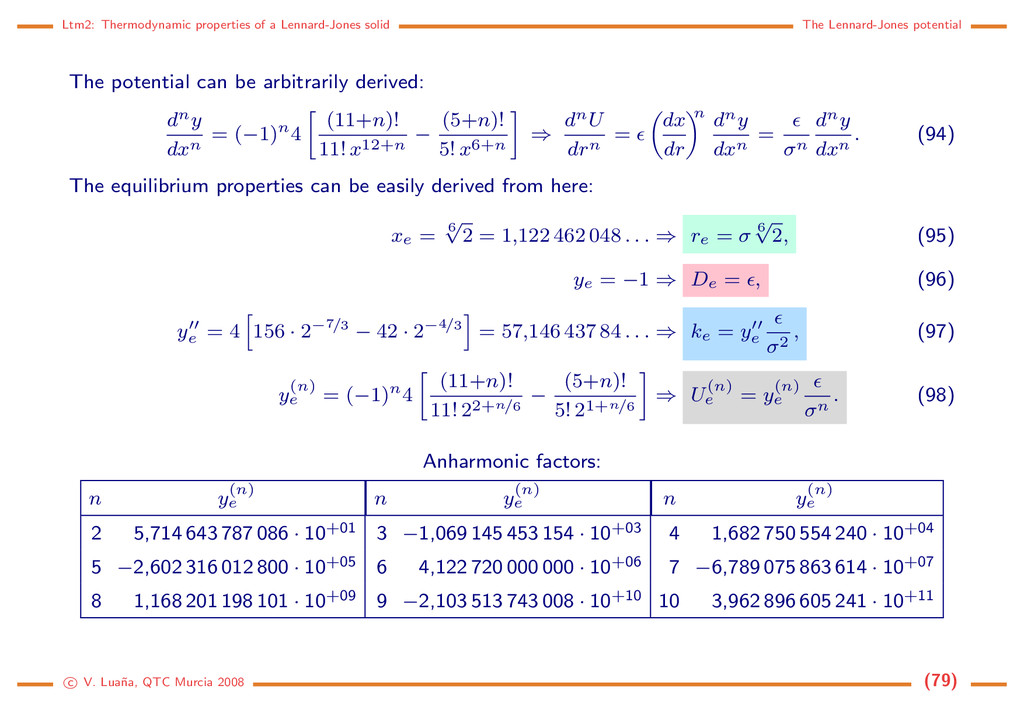{kind=link}
{kind=link}
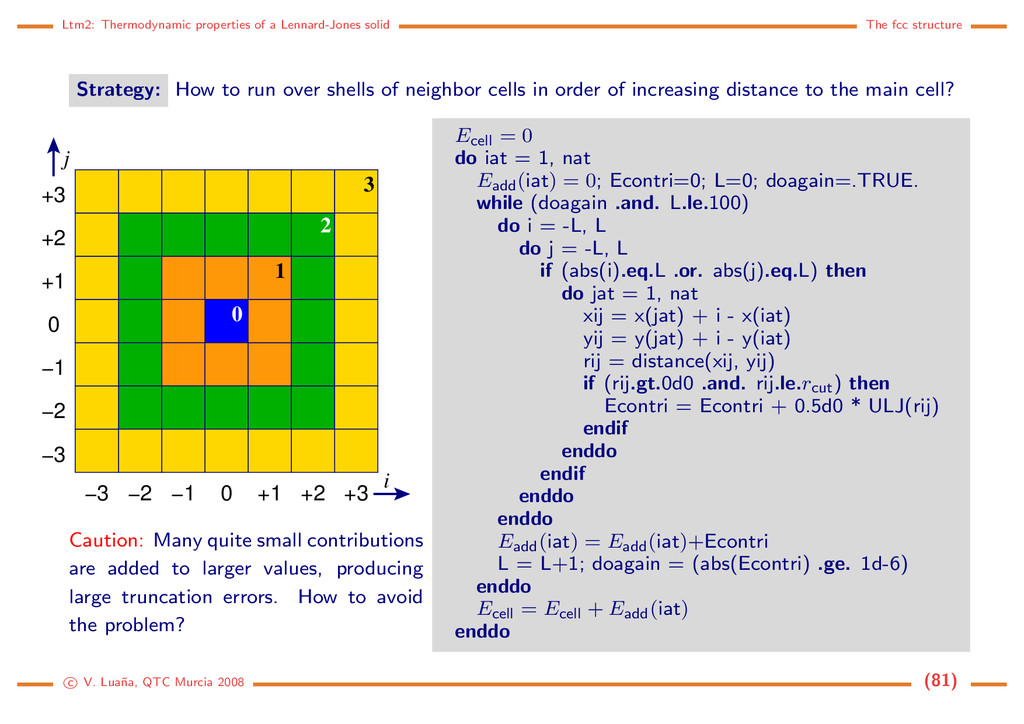{kind=link}
{kind=link}
{kind=link}
{kind=link}
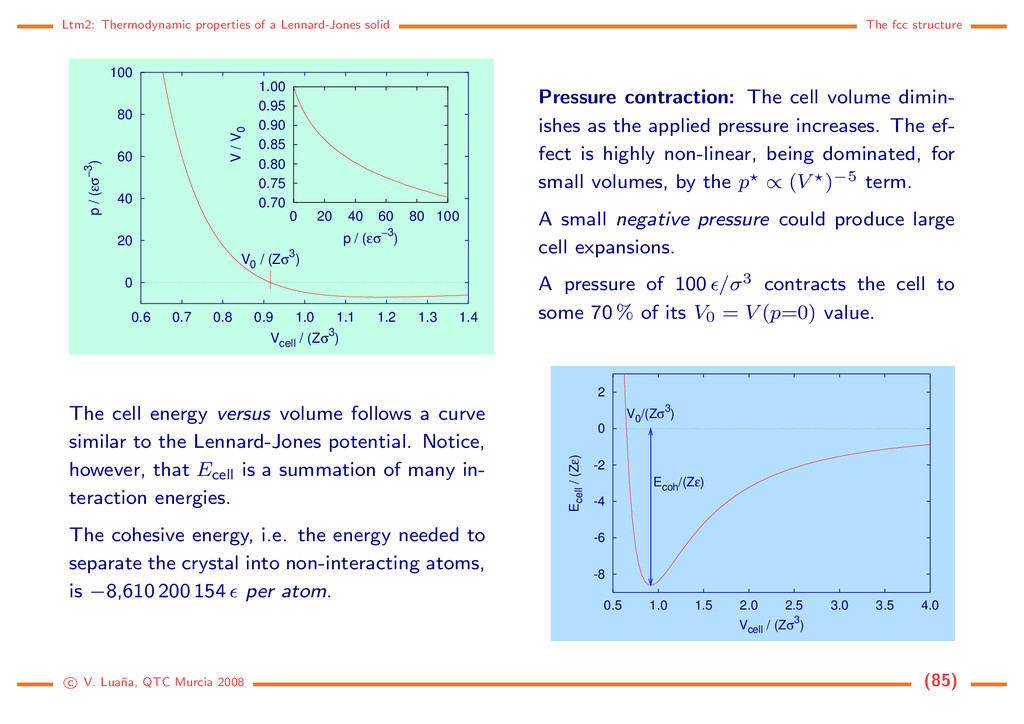{kind=link}
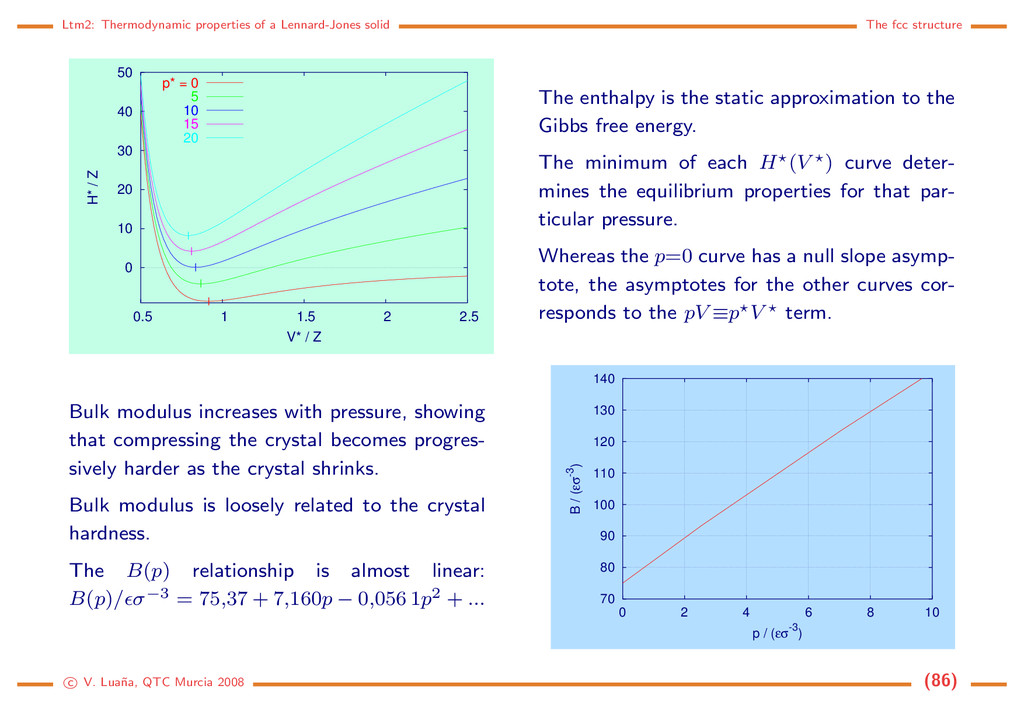{kind=link}
{kind=link}
{kind=link}
{kind=link}
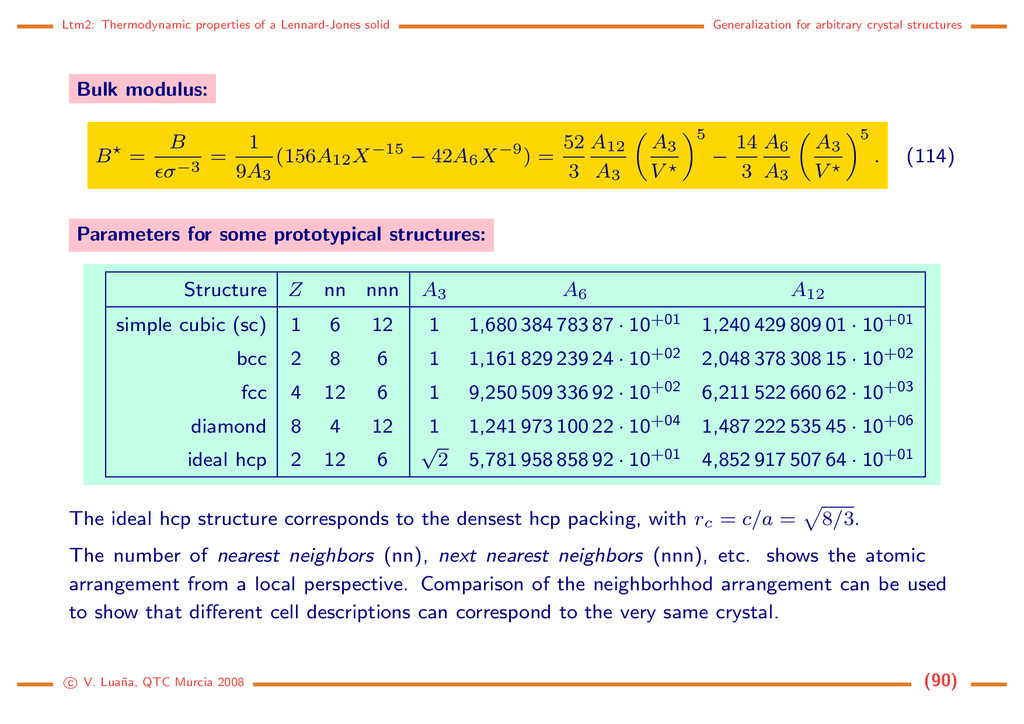{kind=link}
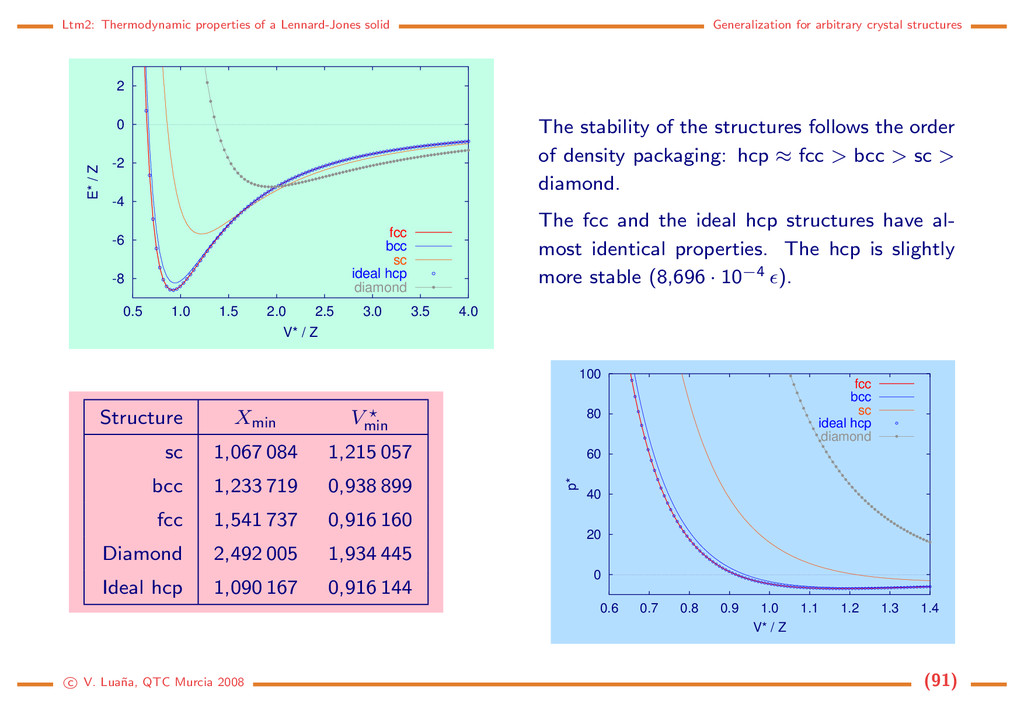{kind=link}
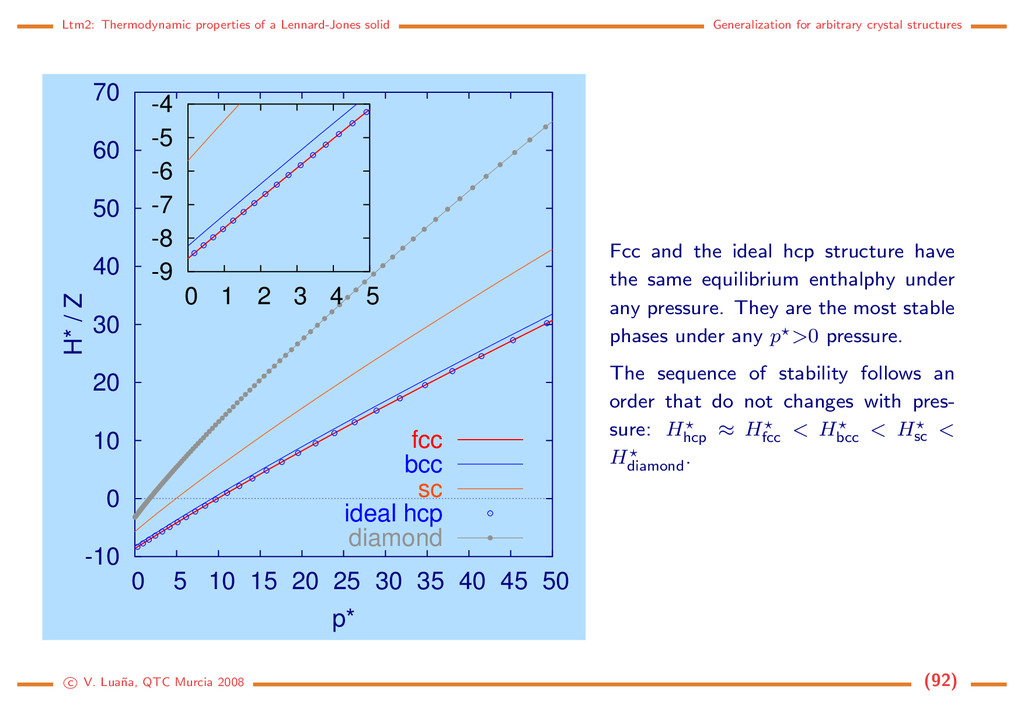{kind=link}
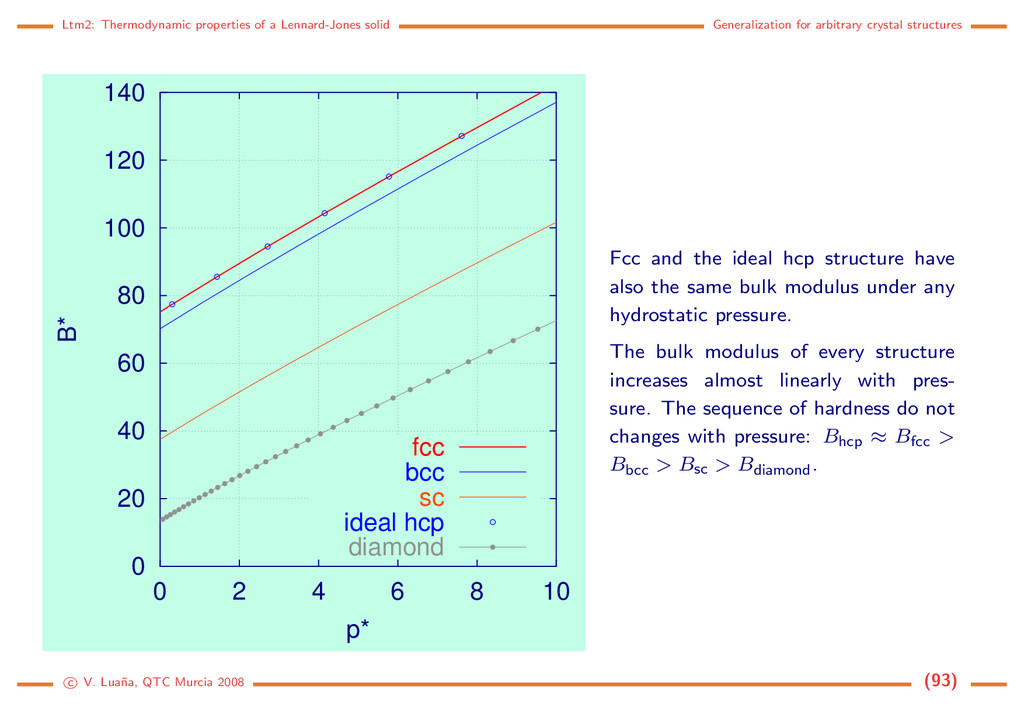{kind=link}
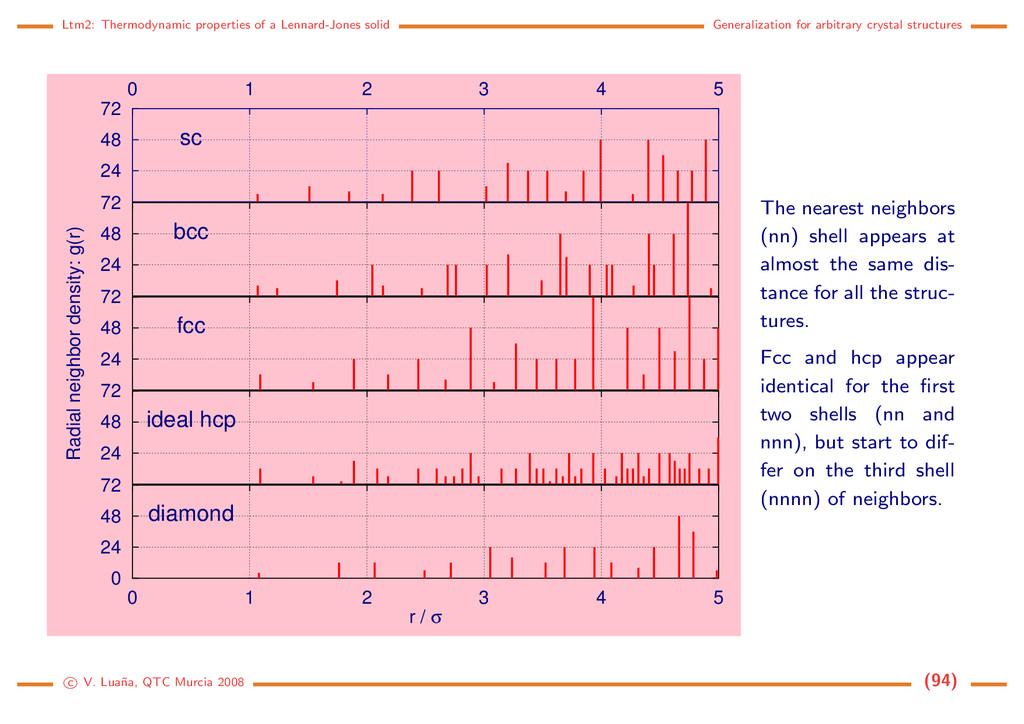{kind=link}
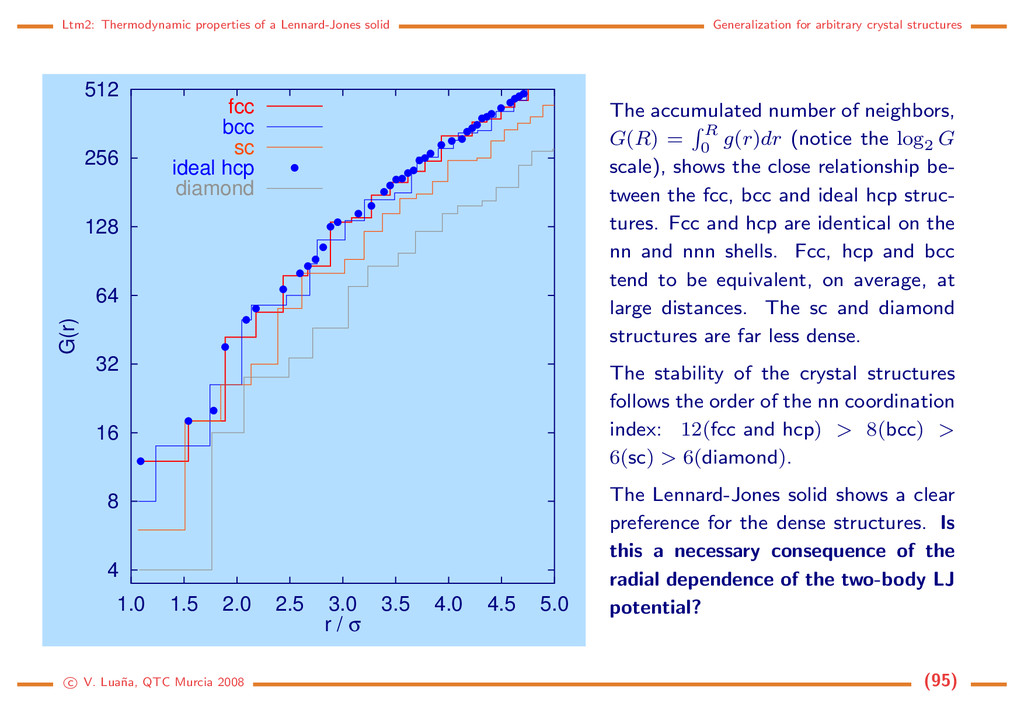{kind=link}
{kind=link}
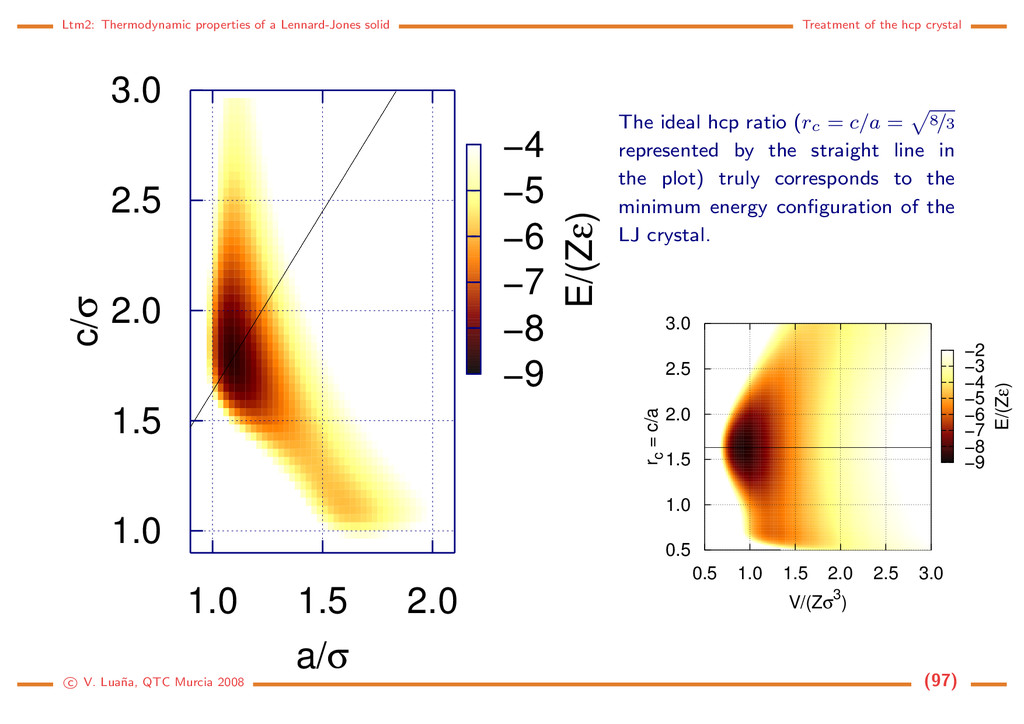{kind=link}
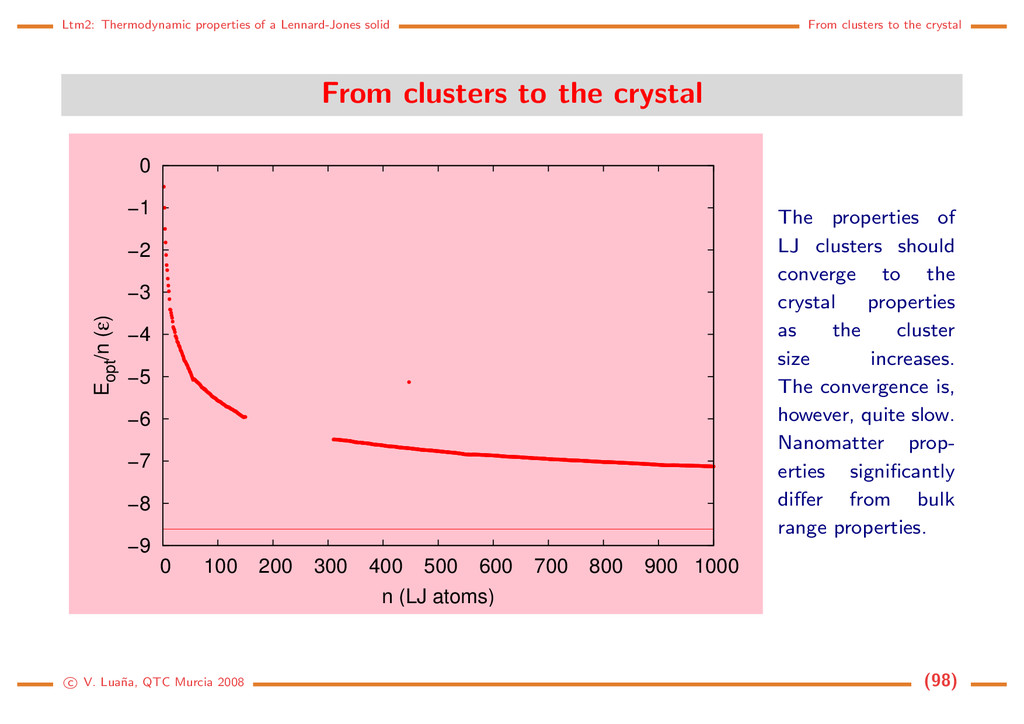{kind=link}
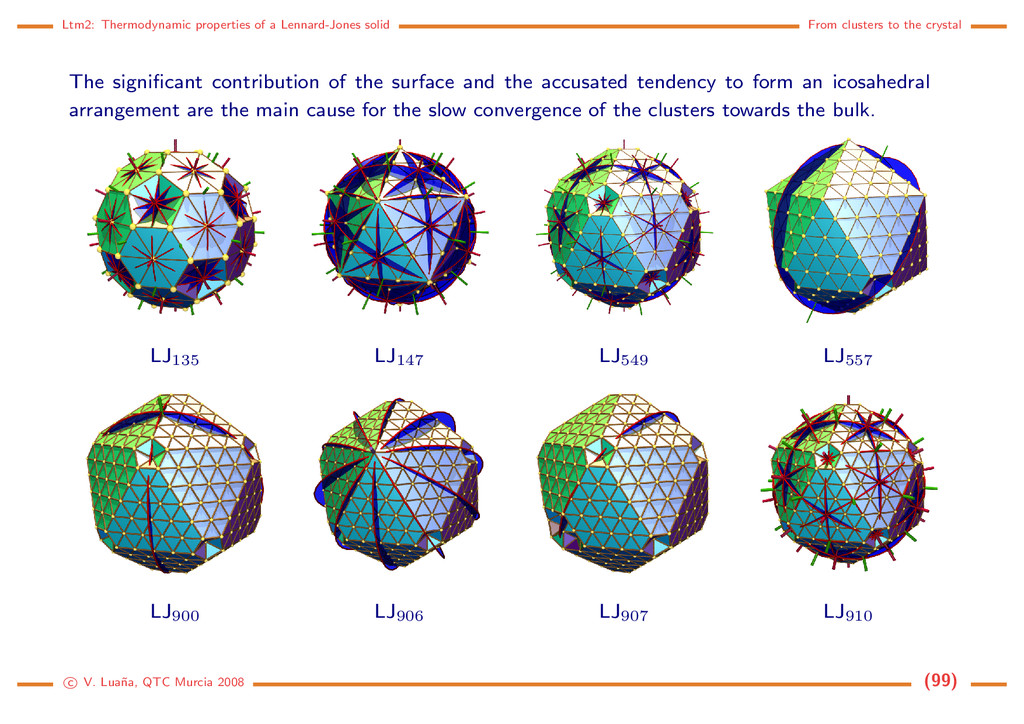{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
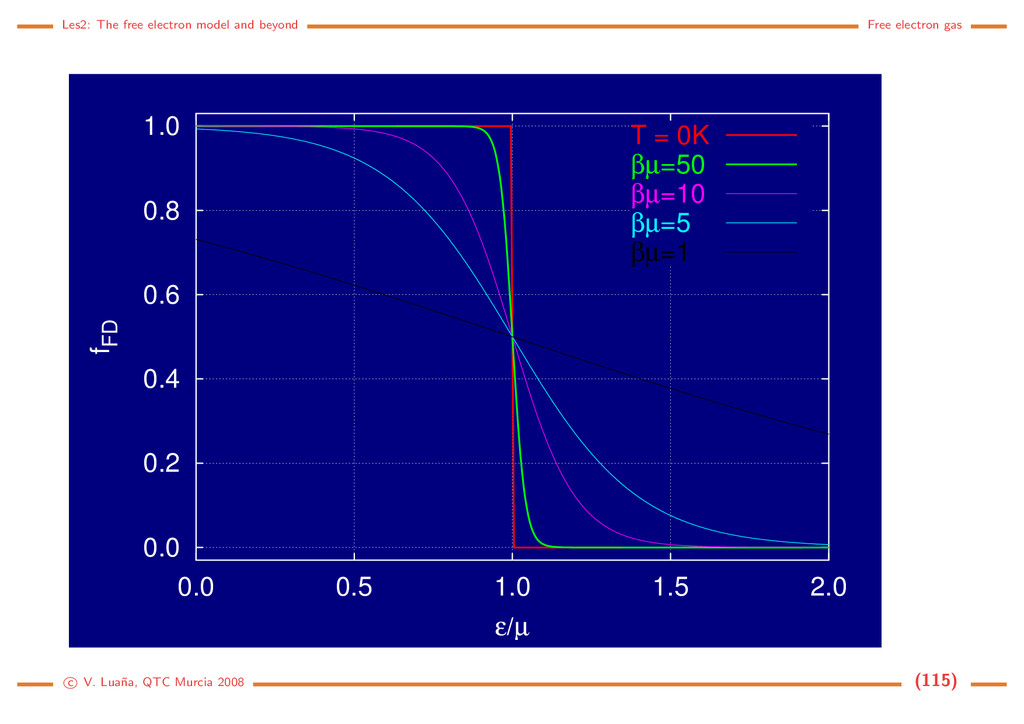{kind=link}
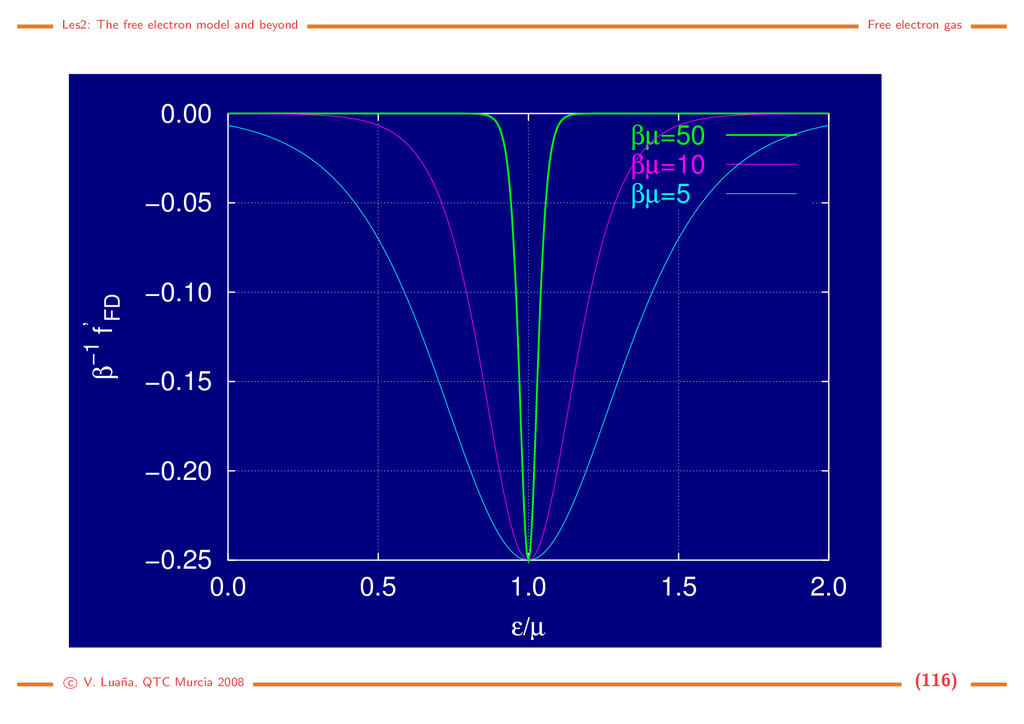{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
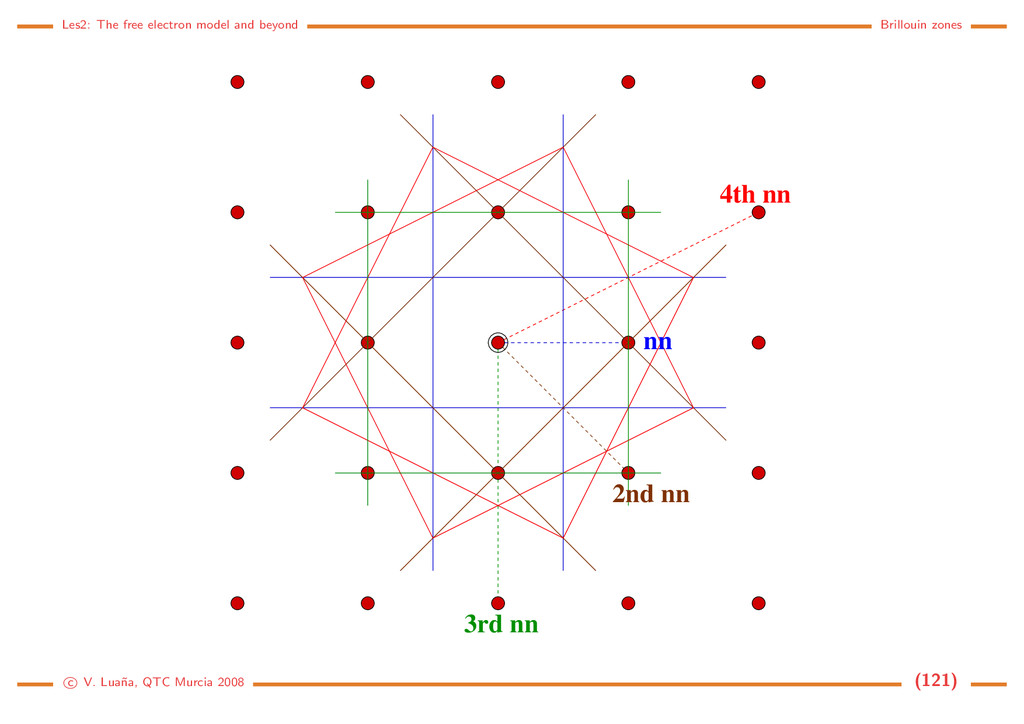{kind=link}
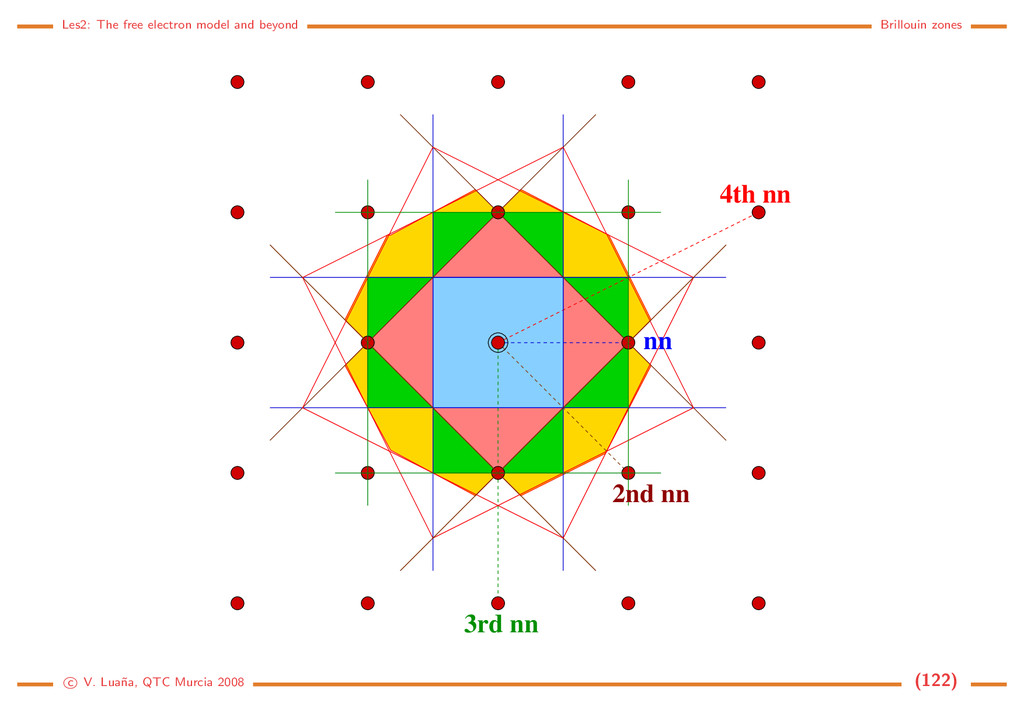{kind=link}
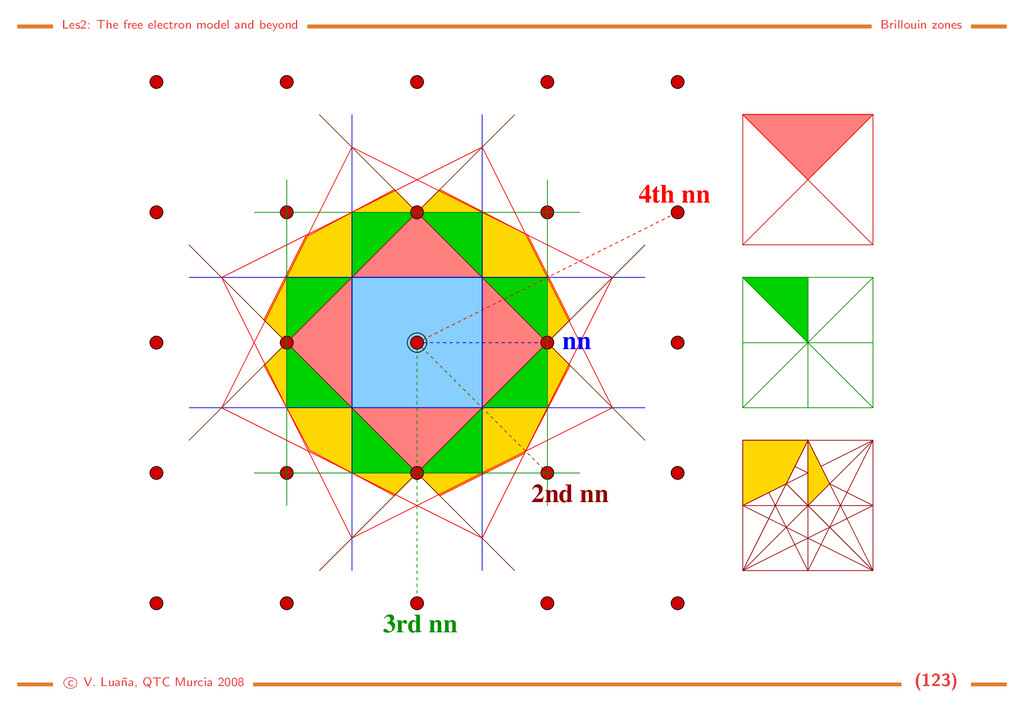{kind=link}
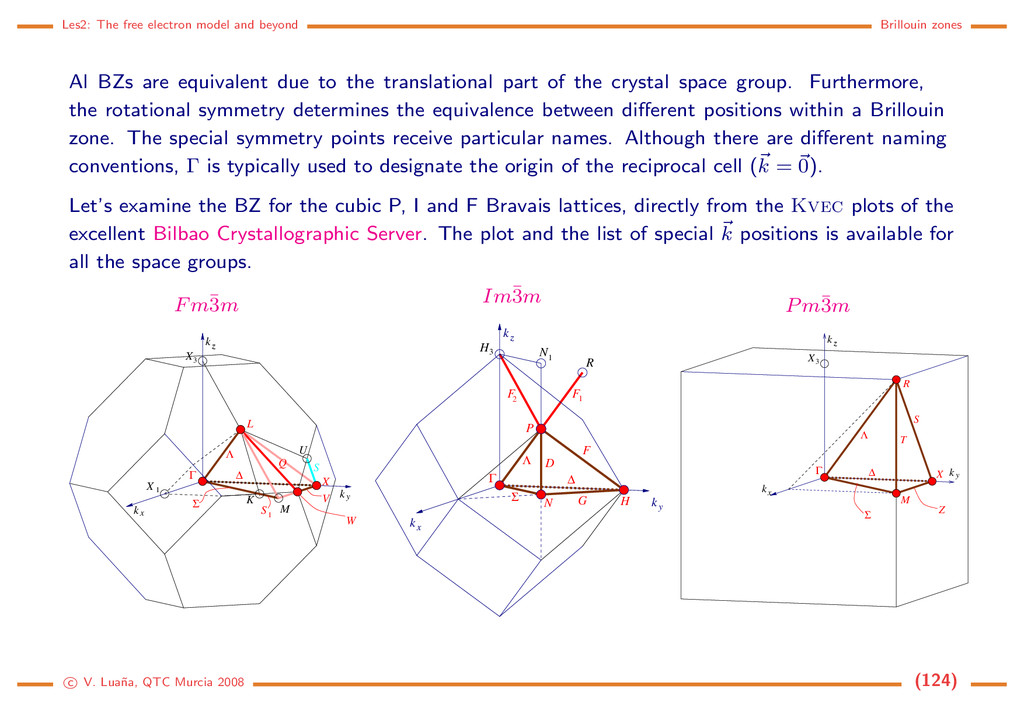{kind=link}
{kind=link}
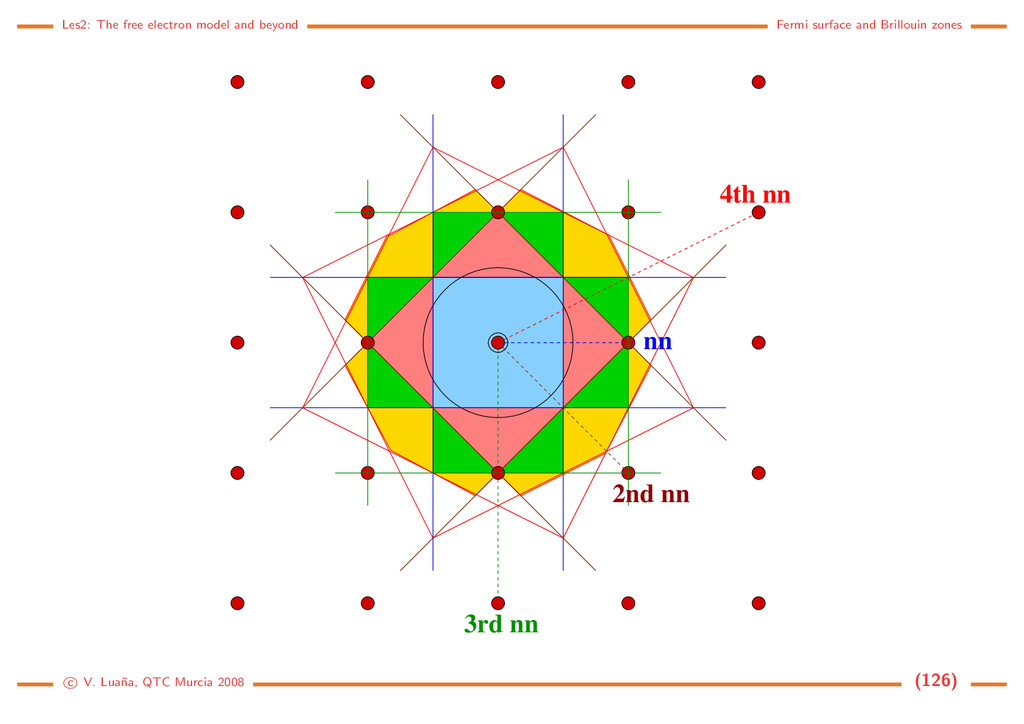{kind=link}
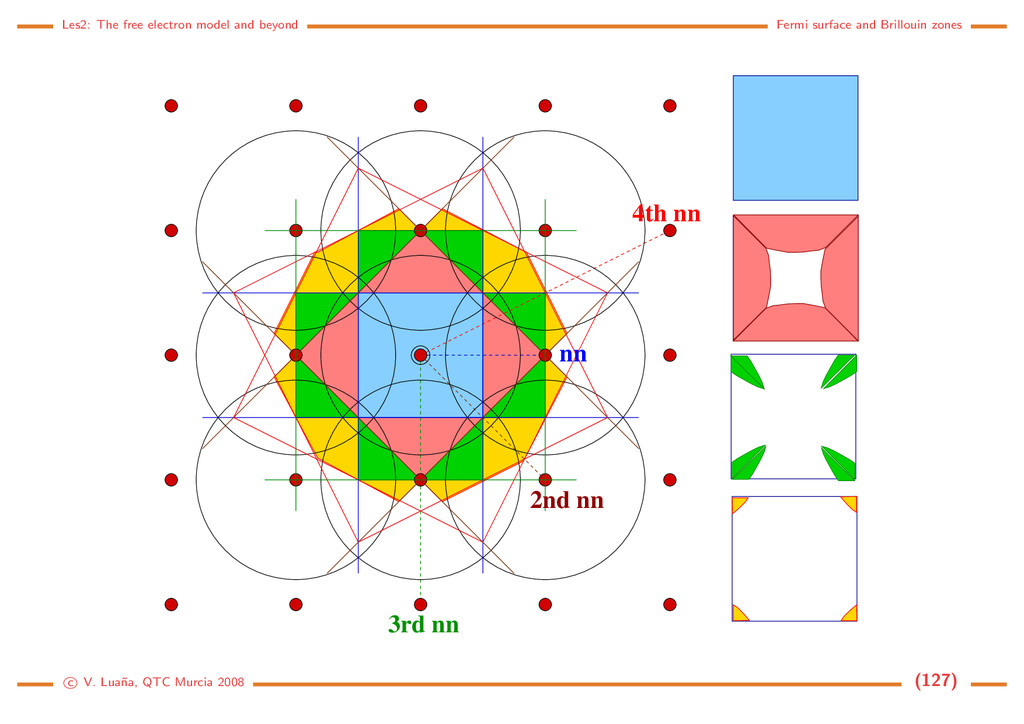{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
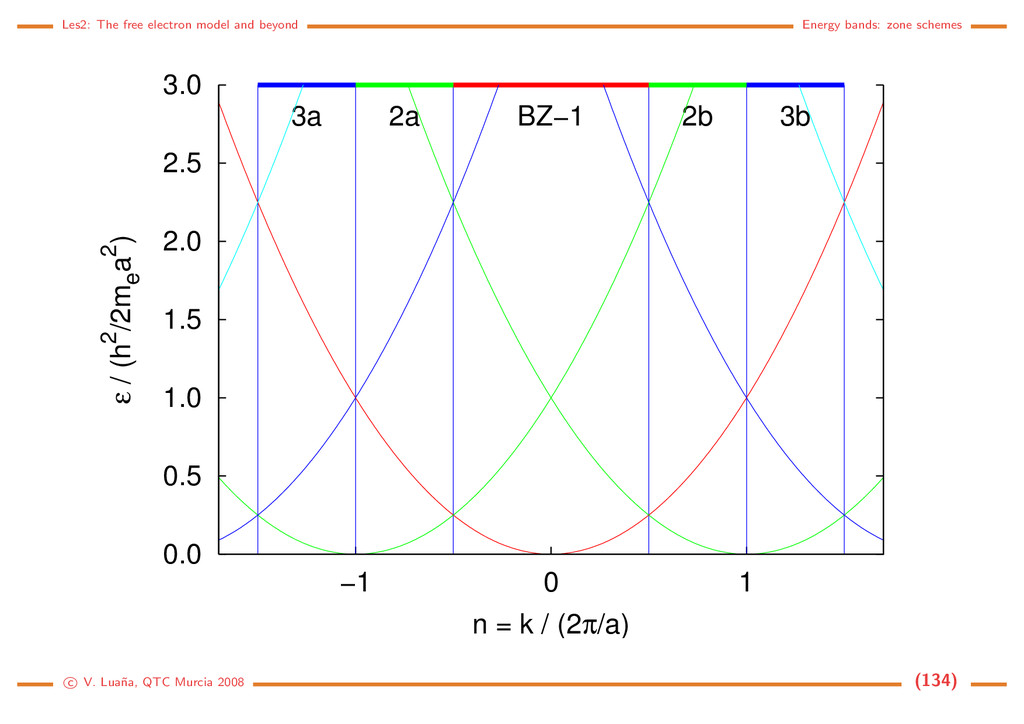{kind=link}
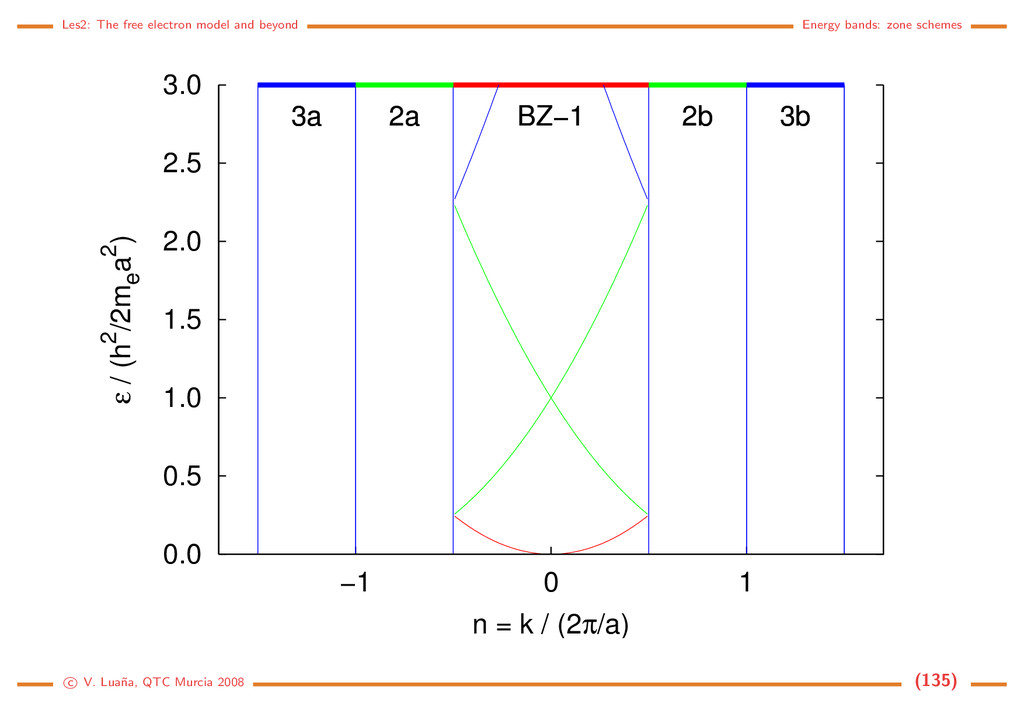{kind=link}
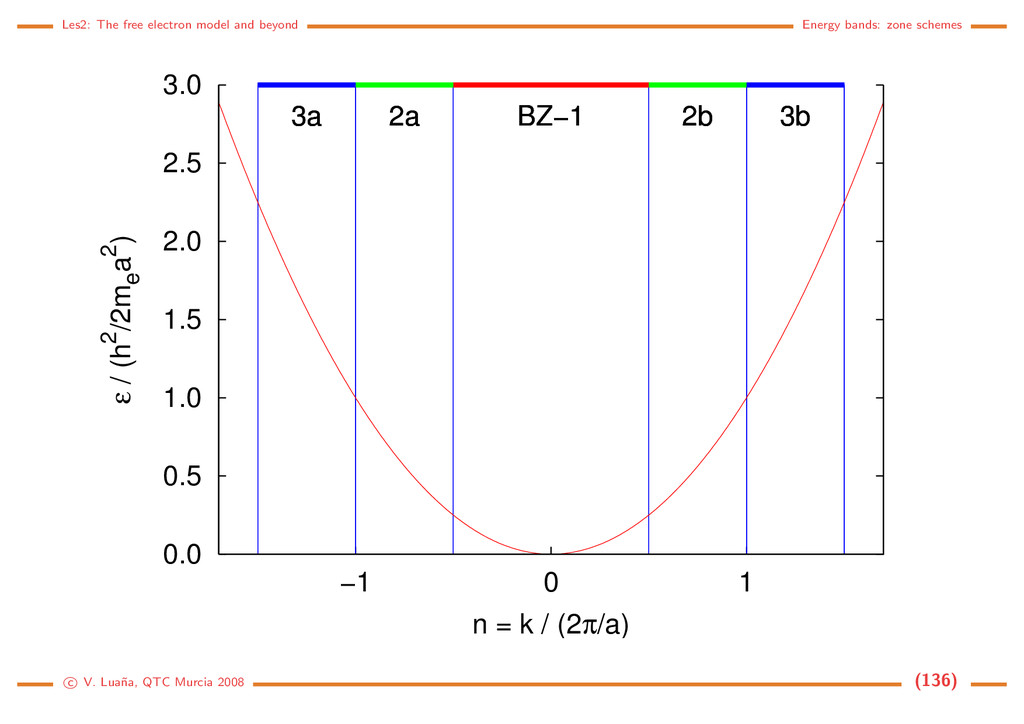{kind=link}
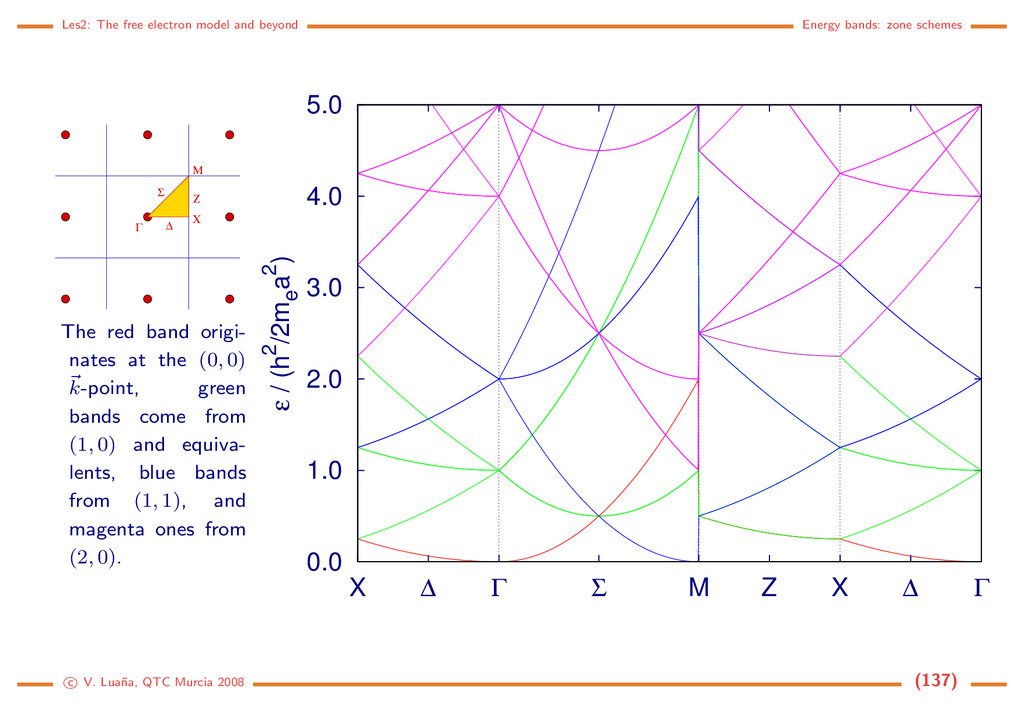{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
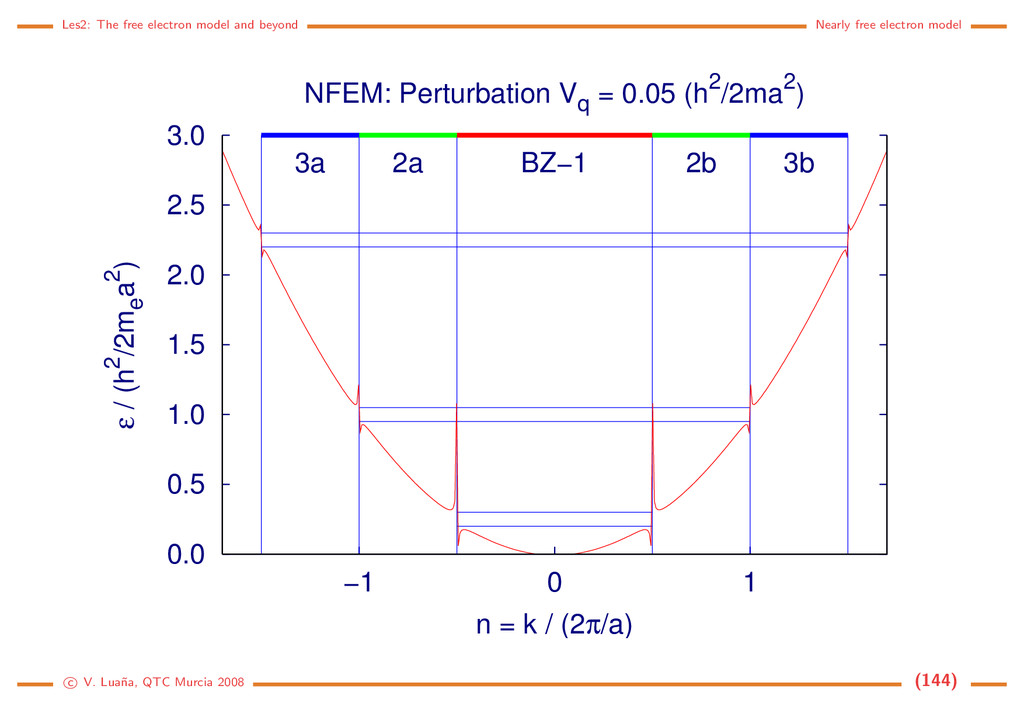{kind=link}
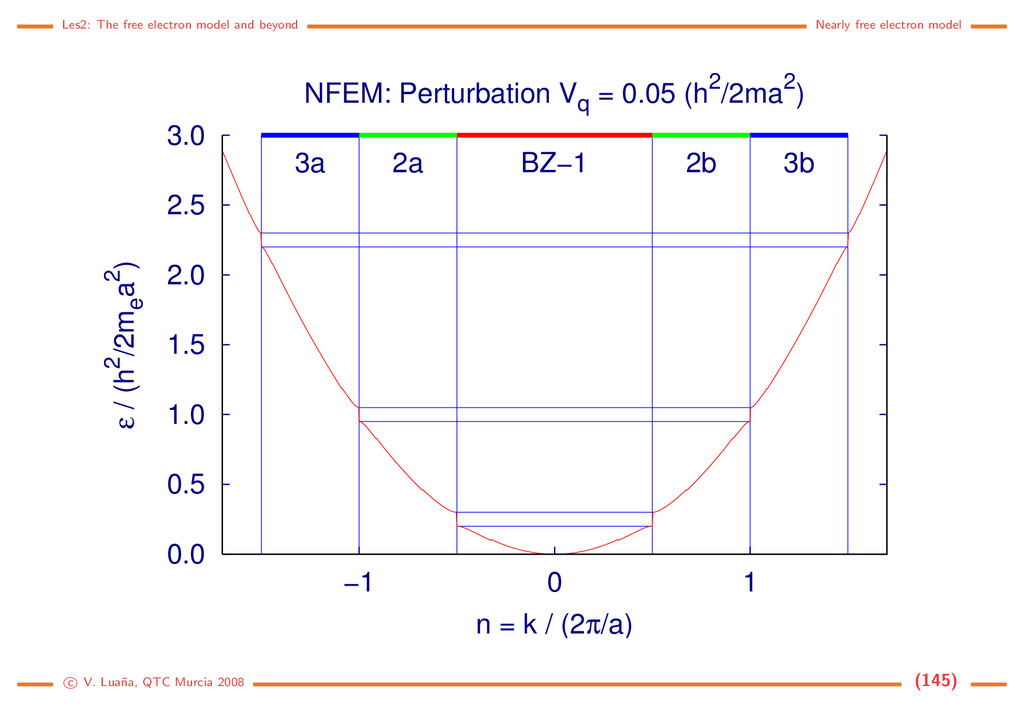{kind=link}
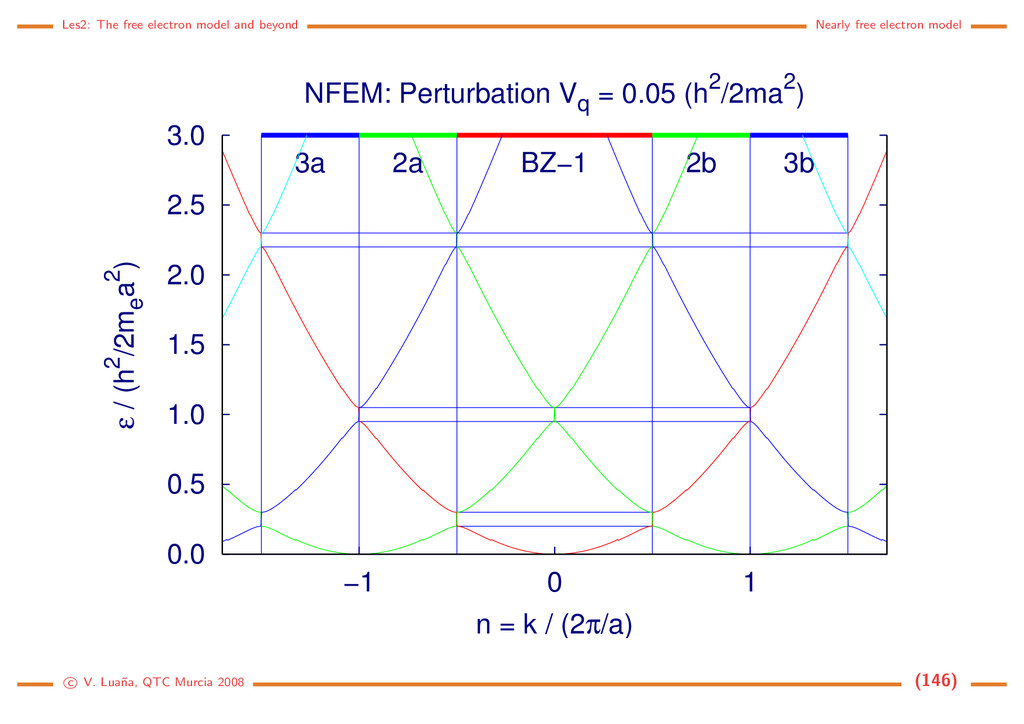{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
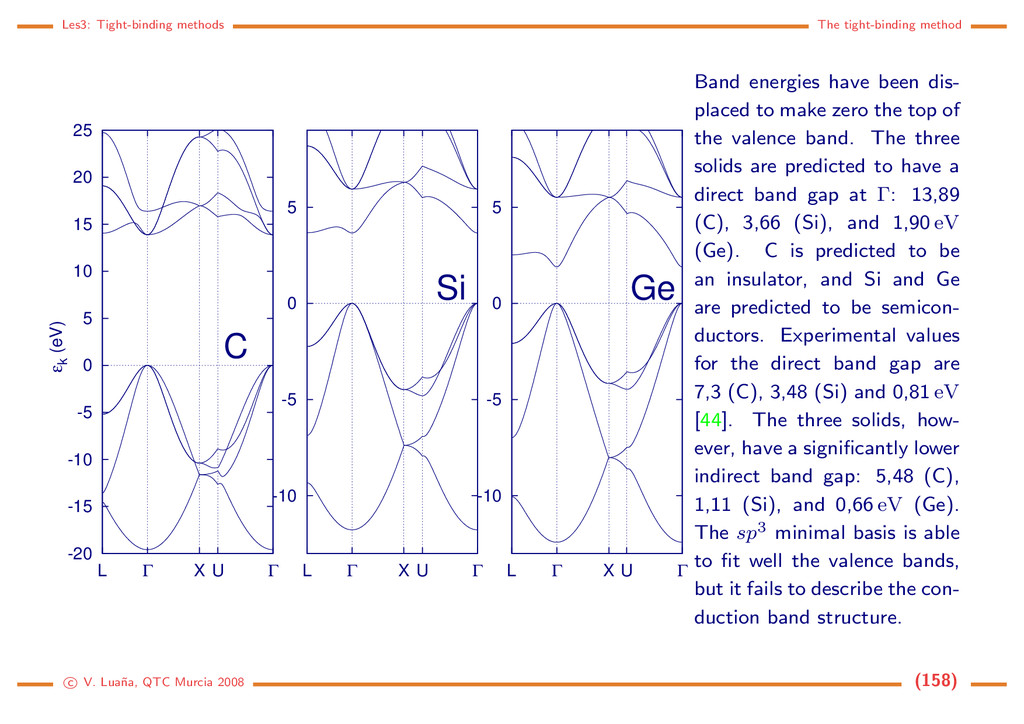{kind=link}
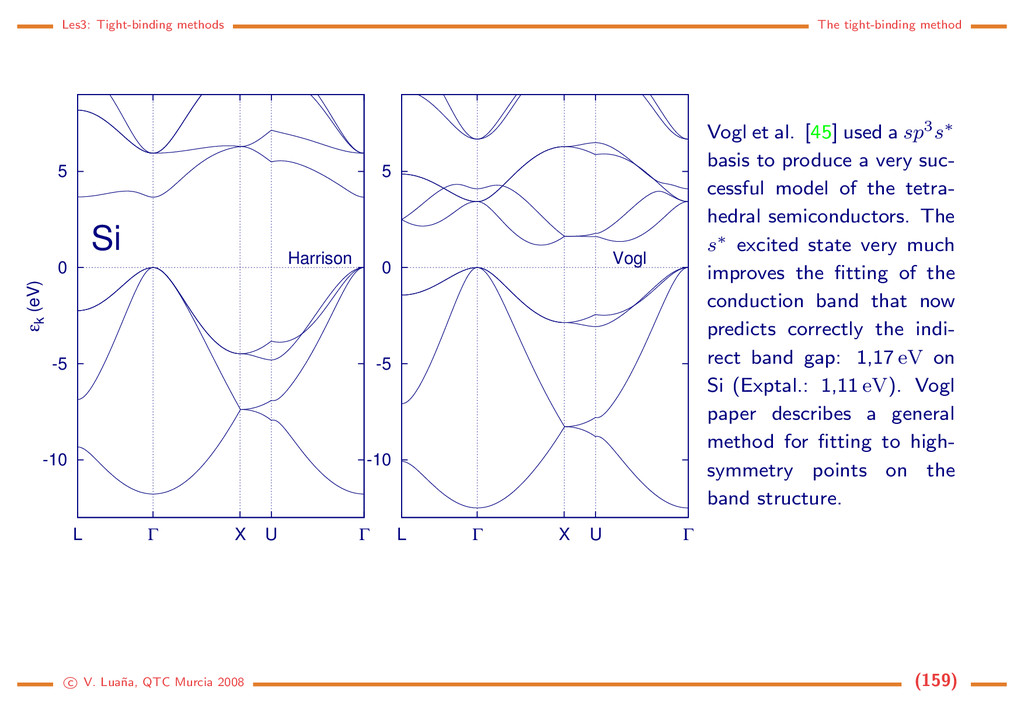{kind=link}
{kind=link}
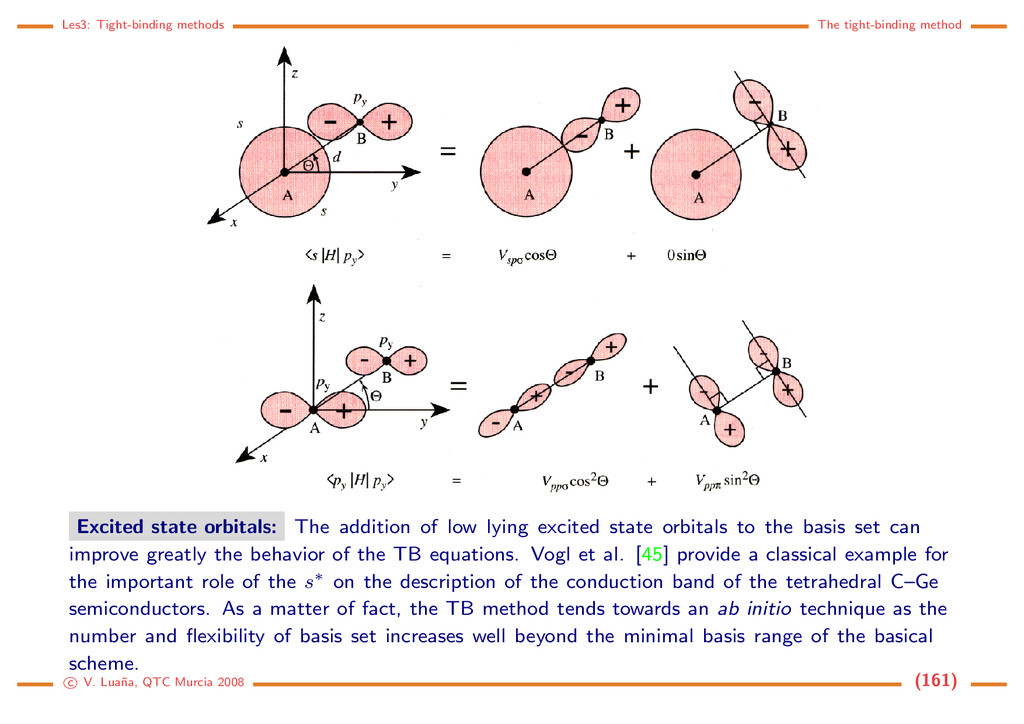{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
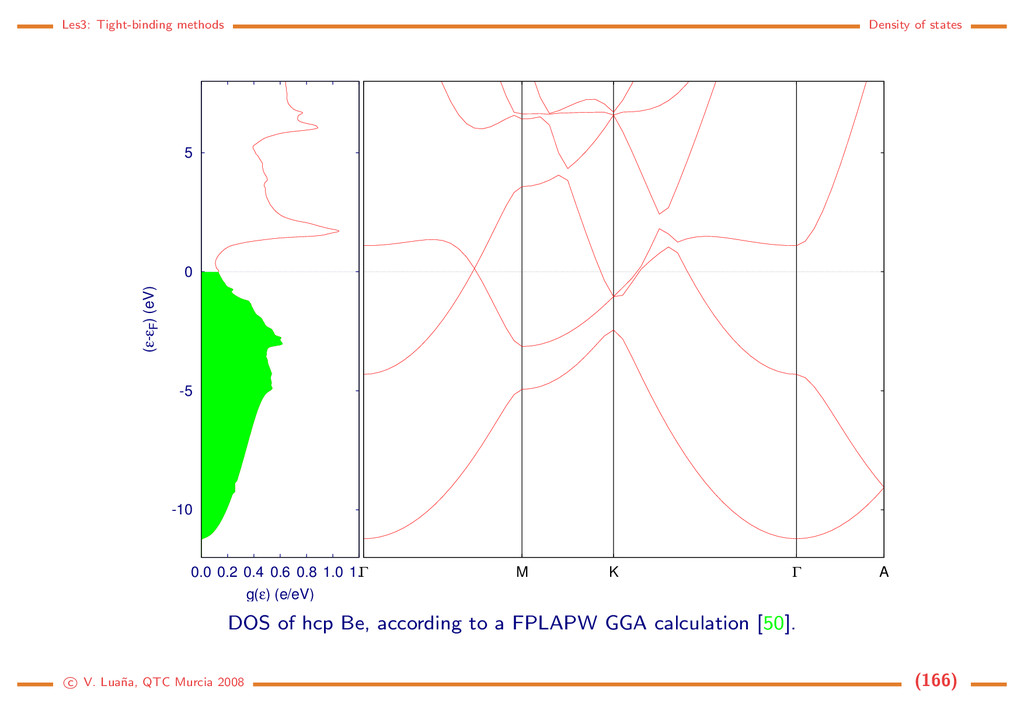{kind=link}
![Les3: Tight-binding methods Exercises References Ashcroft-Mermin, 1976 [1]; Harrison, 1989](https://files.speakerdeck.com/presentations/e45d9cf068ec013133b97691c1dda2be/slide_166.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
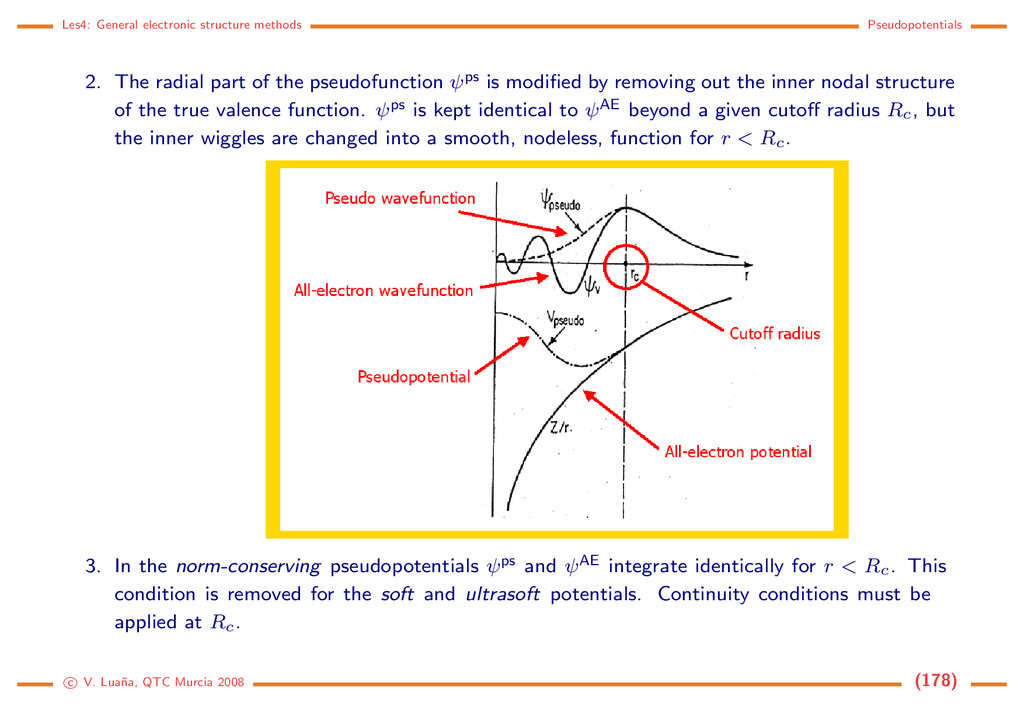{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![References References References [1] N. W. Ashcroft and N. D.](https://files.speakerdeck.com/presentations/e45d9cf068ec013133b97691c1dda2be/slide_183.jpg){kind=link}
![References References [18] V. Lua˜ na and M. Fl´ orez,](https://files.speakerdeck.com/presentations/e45d9cf068ec013133b97691c1dda2be/slide_184.jpg){kind=link}
![References References [39] L. Pueyo Casaus, Estructura electr´ onica de](https://files.speakerdeck.com/presentations/e45d9cf068ec013133b97691c1dda2be/slide_185.jpg){kind=link}