textbooks Electronic structure of solids: quantum espresso Víctor Luaña (†) & Alberto Otero-de-la-Roza (‡) (†) Departamento de Química Física y Analítica, Universidad de Oviedo (‡) Universidad de California at Merced European school on Theoretical Solid State Chemistry ZCAM, Zaragoza, May 13–17, 2013 V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 1 / 86
textbooks Hartree-Fock and Kohn-Sham equations Hartree-Fock (closed shell) Orbital approximation to solve the non-relativistic electronic stationary states: ˆ HeΨ({xi}; {Rα}) = EΨ ⇒ ˆ Fψλ = λψλ (1) − 1 2 ∇2 r + N α Zα riα + R3 ρ(r dr ) |r − r | +ˆ VX ψλ(x; {Rα}) = λ({Rα})ψλ(x; {Rα}) (2) where ˆ VXψλ = − occ. i R3 ψ∗ i (r )ψλ(r ) |r − r | dr ψi(r) (3) is the Hartree (exact) exchange acting over orbital ψλ. SCF: both, HF and KS equations must be solved iteratively until convergence (self-consistency). Kohn-Sham Orbital ansatz to solve the density functional non-relativistic electronic ground state: ˆ HeΨ0({xi}; {Rα}) = EΨ0 ⇒ ˆ Fψλ = λψλ (4) − 1 2 ∇2 r + N α Zα riα + R3 ρ(r )dr |r − r | +ˆ Vxc ψλ(x; {Rα}) = λ({Rα})ψλ (5) where ˆ Vxc is the unknown exchange and correlation functional. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 4 / 86
textbooks Quantum Chemistry and Solid State formalisms I Quantum Chemistry Objective: wavefunctions and total energies of any stationary state. Born-Oppenheimer: separate nuclear and electronic motions to simplify. Nonrelativistic: use classical kinetic energy, separate spin and orbital parts, ... Orbitals: way to produce antisymmetric many electron wavefunctions, via Slater determinants. HF orbitals fulfill Koopmans and Brillouin theorems. HF closed or open shell: the main method to produce orbitals: optimize the orbitals by minimizing the energy of an electronic configuration under the condition that the spinorbitals remain orthonormal. Other methods to produce orbitals: UHF (unrestricted HF), GVB (generalized Valence Bond), MCSCF (Multiconfigurational SCF), ... Basis sets (molecular systems): primitive gaussians (GTO), usually combined to form con- tracted GTOs. Pople family: STO-3G, 3-21G, 6-311++G**, ... Dunning family: cc-PVDZ, cc-PVTZ, cc-PVQZ, aug-cc-PVDZ, ... CBS: Complete Basis Set (extrapolation). techniques: HF assumes an average interaction between electrons. The difference to the exact solution is the correlation problem. Correlation techniques: Möller-Plesset (Many Body Perturbation Theory: MP2, MP3, ...), Configuration Interaction (CIS, CISD, CISDT, ...), Coupled Cluster (CCSD, CCSDT, ...), ... Full CI. lim →CBS,→FCI calculation = exact V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 5 / 86
textbooks Quantum Chemistry and Solid State formalisms II Size scaling (N: spinorbitals, electrons) method lin.† HF, KS HF, KS‡ KS∗ MP2 MP3 CCSD CCSD(T)Q FCI O N N3 N4 N5 N6 N7 N10 N! † Special linear versions of HF and KS methods. ‡ KS with LDA, LSDA, or GGA xc functionals. ∗ KS with hybrid functionals. Solid State (Density Functional) Objective: electron density and total energy of the ground state. Born-Oppenheimer: Nonrelativistic formalism: most solid state codes include relativistic corrections. Different types of basis set formalisms: (1) planewaves and pseudopotentials (pw+ps); (2) GTO’s; (3) local orbitals and ps.; (4) FPLAPW (Full Potential Linear Augmented PlaneWaves); ... xc functionals: Metaphorical classification (Jacob’s Ladder): (1) LDA/LSDA [Ex: VWN91]; (2) GGA [Ex: PBE, PW91]; (3) meta-GGA [Ex: TPSS]; (4) hybrid [Ex: B3LYP]; (5) double hybrid [Ex: XYG3]; (¿?) the unknown exact functional. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 6 / 86
textbooks KS equations on a crystal (quantum espresso version) −∇2 r + α Vα sf + VH(r) + Vxc(r) ψnk(r) = n(k)ψnk(r) (6) −∇2 r : kinetic energy, Rydberg units are used throught. Vα sf : non-local pseudopotential. Either norm-conserving, ultrasoft, or PAW (projected aug- mented wave) pseudopotentials can be used. VH(r) = ∂EH[ρ] ∂ρ = R3 ρ(r dr |r − r | : Coulomb potential or Hartree term. Vxc(r) = ∂Exc[ρ] ∂ρ : exchange and correlation potential. Bloch states: ψnk(r) = eikrunk(r), where ψnk(r) is periodical over reciprocal space and unk(r) is periodical over real space. ρ(r) = nk fnk |ψnk|2 = nk fnk |unk|2 , with fi ∈ {0, 1} (a typical insulator) or fi = 1/(1 + e− i/kT ) (Mermin functional, used on metals). k ∈ BZ1. Running special points and directions in iBZ1 (irreducible first Brillouin zone) is the basis for band diagrams. Total properties (energy, DOS, etc) requires integration of BZ1. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 8 / 86
textbooks KS calculations on a basis Let φ q+G (r) be a basis function φq (r) at the reciprocal cell G. The basis is used to build the KS orbitals: ψnq (r) = nq cnq (G)φ q+G (r). (7) By minimizing the energy with cnq (G) as the variational parameters the KS equations transform into ∀ G G H q+G,q+G − nqS q+G,q+G cnq (G) = 0, (8) that must be solved for every q ∈ BZ1. In the above equation: H q+G,q+G = φ q+G (r) − 1 2 ∇2 + V φ q+G (r) , S q+G,q+G = φ q+G φ q+G , (9) and the KS equation takes the form of a generalized eigen equation HC = SCE that must besolved for every q. By sampling q-points along special directions on the BZ1 we get the band diagrams: nq . The total energy (E), Density of States (DOS, g(E)), and the Fermi surface in the case of a metal ( F(q)), are produced by integration for all q ∈ BZ1. Mokhorst-Pack special points methods is popular for doing this integration. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 9 / 86
textbooks Planewaves (3D) and the movement of a free electron I The hamiltonian is ˆ h = ˆ p2/2m, ˆ p = −i ∇, and the solutions are planewaves (PW) again: |k = ψ k (r ) = V−1/2eik·r, k = 2k2 2m , r, k ∈ R3. (10) with V being the volume of the normalization box. Some properties The PWs are eigenfunctions of the momentum operator: ˆ p |k = k |k . The particle velocity is proportional to k, the wavevector : v = k/m. The |k and |−k PWs are degenerated. The wavelength of a PW is λ = 2π/k. To enforce periodic boundary conditions in a general way we define the parallelepipedic cell a ˜ and an arbitrary primitive translation t = a ˜ n, n ∈ Z3. For any r ∈ R3: ψ k (r + t ) = ψ k (r ) =⇒ eik·t = 1. (11) If k is a vector in the reciprocal cell, k = a ˜ k = 2πa ˜ h, the periodicity condition is 1 = eik·t = ei2π(h Tn) =⇒ h1nx + h2ny + h3nz ∈ Z for all n ∈ Z3. (12) V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 10 / 86
textbooks Planewaves (3D) and the movement of a free electron II The wavevector of the periodic PWs is then k = 2π(h1a + h2b + h3c ) = 2πa ˜ h = a ˜ k, (h1, h2, h3) ∈ Z3. (13) This discrete mesh of allowed k-points is distributed uniformly in the reciprocal space. Each k point can be associated with a small parallelepiped of volume v k = (2π)3V = (2π)3/V, where V is the volume of the main cell. It is implicitely assumed that a primitive cell is used to describe the reciprocal space. The periodic PWs form an orthonormal set k|k = 1 V V ei(k−k )rdr = δ k,k , (14) where the integral is done on a unit cell. The set is also complete, which means that any 3D function can be expanded as f(r ) = k f k eik·r ⇐⇒ f k = V f(r )e−ik·rdr. (15) V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 11 / 86
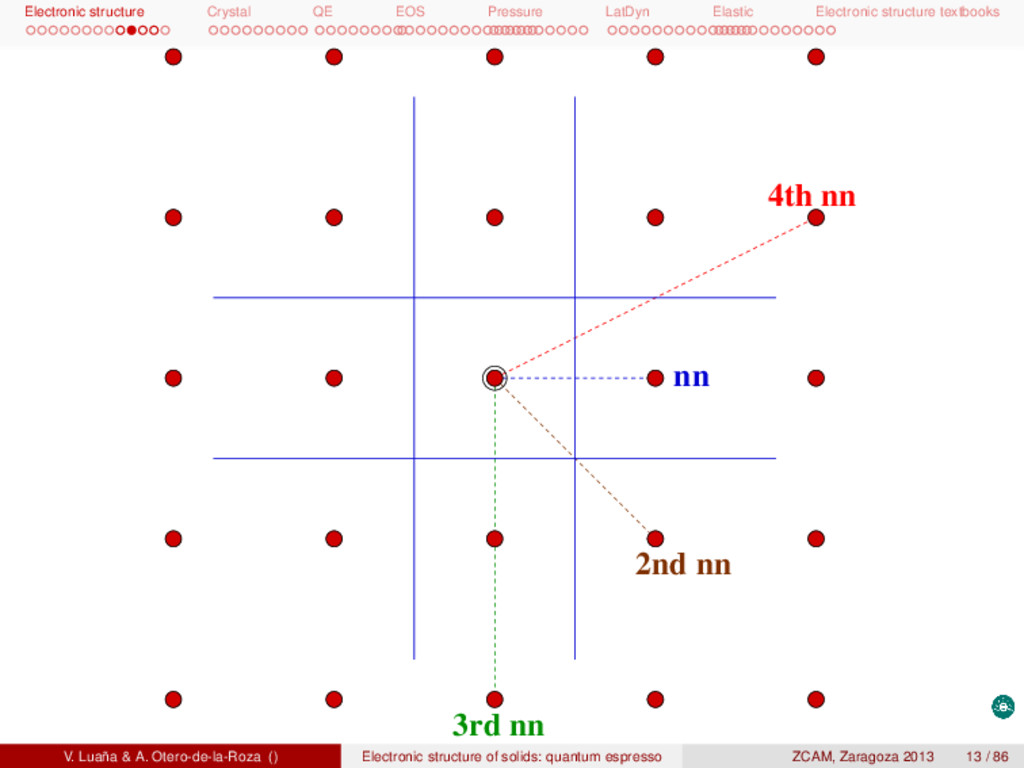
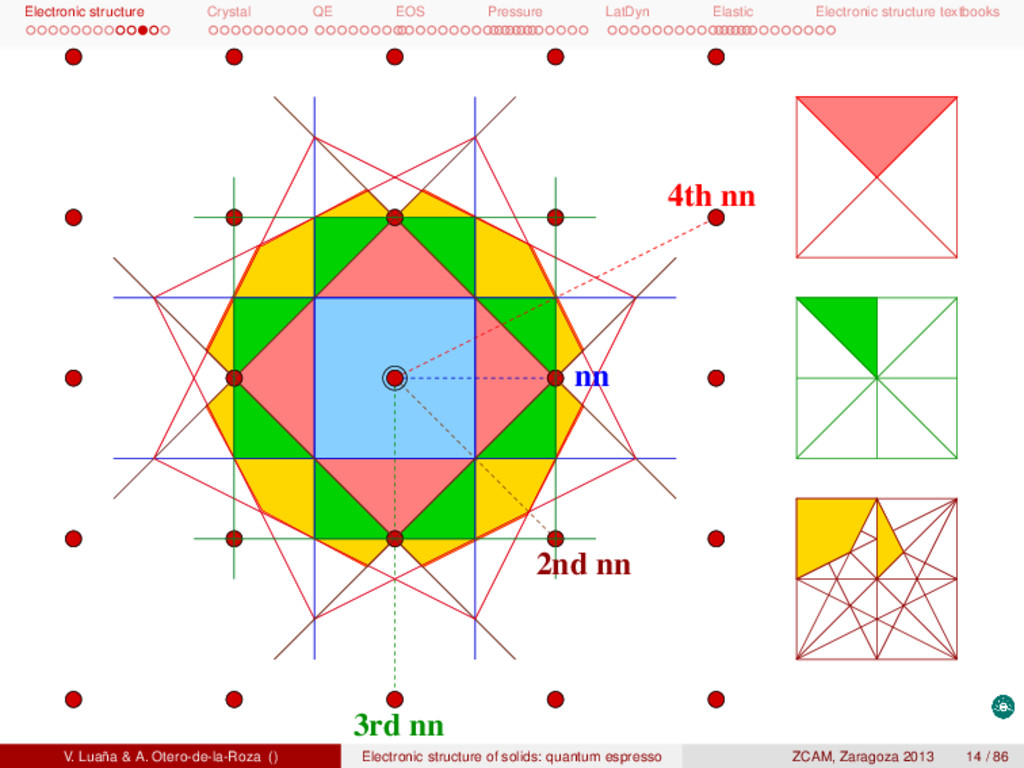
textbooks Brillouin zones I The first Brillouin zone (BZ-1) is the common name for the Wigner-Seitz primitive cell of the reciprocal or k-space lattice. Whereas primitive and centered cells are both used for the direct lattice, centering is avoiding in the manipulation of the k lattice. An alternative definition for BZ-1 is the set of points in k space that can be reached from the origin (k = 0) without crossing any Bragg plane. A Bragg plane for any two points in the lattice being the plane which is perpendicular to the line between the two points and passes through the bisector of that line. The concept of BZ-1 can be generalized. The second BZ (BZ-2) is the set of points that can be reached from the first zone by crossing only one Bragg plane. BZ-(n+1) is formed is the set of points not in {BZ-1, BZ-2, ... BZ-(n−1)} that can be reached from BZ-n by crossing only one Bragg plane. Alternatively, the n Brillouin zone can be reached from the origin by crossing n−1 Bragg planes, but not fewer. The construction of the BZ is illustrated in the next slides for a simple square lattice. An important point to check is that every Brillouin zone has the same volume, namely the volume of a primitive reciprocal lattice. In addition, any BZ can be mapped back to the first zone by using just primitive translations, i.e. all the BZ’s are equivalent by translation symetry. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 12 / 86
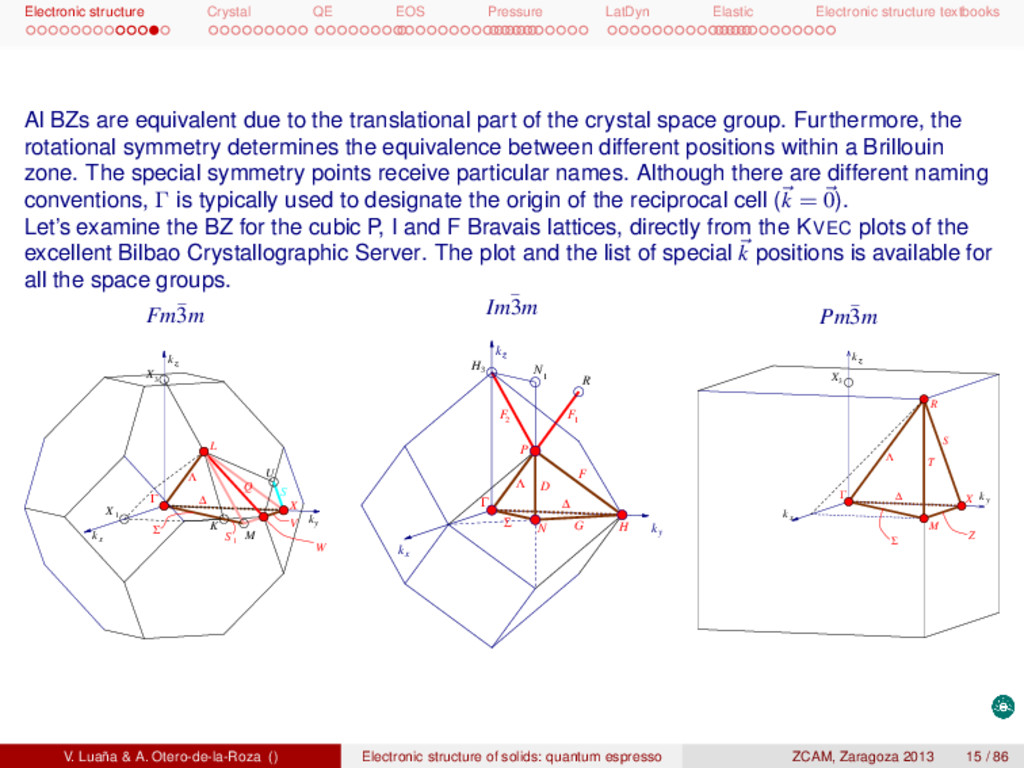
textbooks Al BZs are equivalent due to the translational part of the crystal space group. Furthermore, the rotational symmetry determines the equivalence between different positions within a Brillouin zone. The special symmetry points receive particular names. Although there are different naming conventions, Γ is typically used to designate the origin of the reciprocal cell (k = 0). Let’s examine the BZ for the cubic P, I and F Bravais lattices, directly from the KVEC plots of the excellent Bilbao Crystallographic Server. The plot and the list of special k positions is available for all the space groups. Fm¯ 3m L z kz Γ kx ky W V Σ S 1 3 X Λ ∆ K M Q 1 X U X S Im¯ 3m k k x y z kz H G N Σ Γ ∆ D Λ F P F 1 R N 1 3 H F 2 Pm¯ 3m z kz Γ Λ ky ∆ Z kx X M Σ R T S 3 X V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 15 / 86
textbooks Band diagrams and electronic density of states InN: a direct gap semiconductor V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 16 / 86
textbooks Crystal geometry a c b α γ β Cell parameters: (a, b, c, α, β, γ). Cell vectors matrix: h = (a, b, c)T . Cell volume: V = a · (b × c) = b · (c × a) = c · (a × b). Crystallographic coordinates: ri = hT xi = xia + yib + zic. {x, y, x} ∈ [0, 1) (main cell). Metric tensor: G = hT h. Scalar product: x · y = xT Gy. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 18 / 86
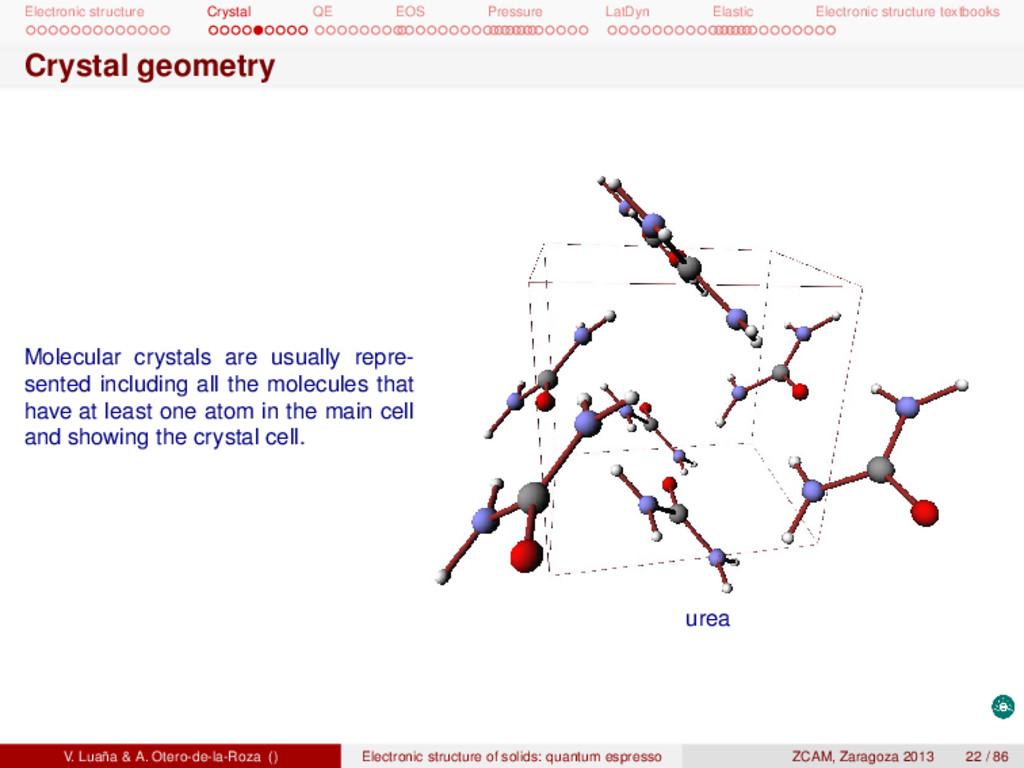
textbooks Crystal geometry Molecular crystals are usually repre- sented including all the molecules that have at least one atom in the main cell and showing the crystal cell. urea V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 22 / 86
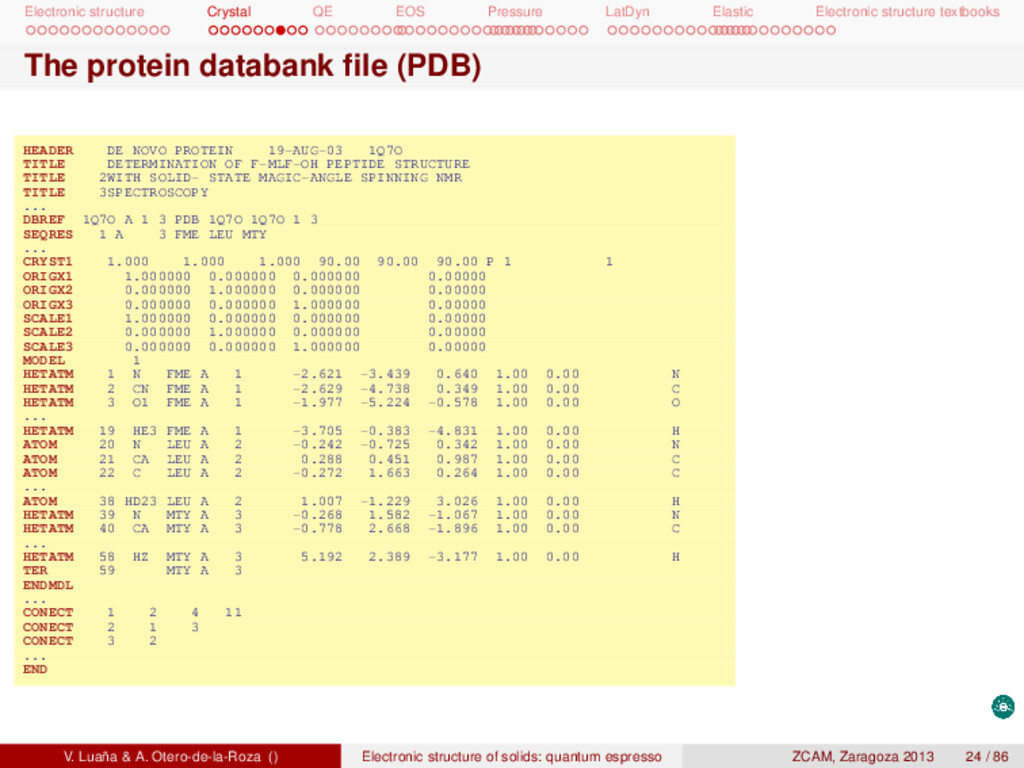
textbooks The protein databank file (PDB) HEADER DE NOVO PROTEIN 19-AUG-03 1Q7O TITLE DETERMINATION OF F-MLF-OH PEPTIDE STRUCTURE TITLE 2WITH SOLID- STATE MAGIC-ANGLE SPINNING NMR TITLE 3SPECTROSCOPY ... DBREF 1Q7O A 1 3 PDB 1Q7O 1Q7O 1 3 SEQRES 1 A 3 FME LEU MTY ... CRYST1 1.000 1.000 1.000 90.00 90.00 90.00 P 1 1 ORIGX1 1.000000 0.000000 0.000000 0.00000 ORIGX2 0.000000 1.000000 0.000000 0.00000 ORIGX3 0.000000 0.000000 1.000000 0.00000 SCALE1 1.000000 0.000000 0.000000 0.00000 SCALE2 0.000000 1.000000 0.000000 0.00000 SCALE3 0.000000 0.000000 1.000000 0.00000 MODEL 1 HETATM 1 N FME A 1 -2.621 -3.439 0.640 1.00 0.00 N HETATM 2 CN FME A 1 -2.629 -4.738 0.349 1.00 0.00 C HETATM 3 O1 FME A 1 -1.977 -5.224 -0.578 1.00 0.00 O ... HETATM 19 HE3 FME A 1 -3.705 -0.383 -4.831 1.00 0.00 H ATOM 20 N LEU A 2 -0.242 -0.725 0.342 1.00 0.00 N ATOM 21 CA LEU A 2 0.288 0.451 0.987 1.00 0.00 C ATOM 22 C LEU A 2 -0.272 1.663 0.264 1.00 0.00 C ... ATOM 38 HD23 LEU A 2 1.007 -1.229 3.026 1.00 0.00 H HETATM 39 N MTY A 3 -0.268 1.582 -1.067 1.00 0.00 N HETATM 40 CA MTY A 3 -0.778 2.668 -1.896 1.00 0.00 C ... HETATM 58 HZ MTY A 3 5.192 2.389 -3.177 1.00 0.00 H TER 59 MTY A 3 ENDMDL ... CONECT 1 2 4 11 CONECT 2 1 3 CONECT 3 2 ... END V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 24 / 86
textbooks Crystal Data Bases I Cambridge Structural Database (CSD) (http://www.ccdc.cam.ac.uk/products/csd/): The main source of crystal structures for organic and organometallic compounds. Commercial, access by subscription. 596810 structures as of 2012-01-01. Inorganic Crystal Structure Database (ICSD) (http://www.fiz-karlsruhe.de/icsd.html?&L=0): Natural and synthetic inorganic compounds. Commercial, access by subscription. 142179 entries (nov 2011). See a fully functional subset at http://icsd.ill.fr/icsd/ (3592 samples). CRYSTMET (http://www.tothcanada.com/databases.htm): Metals, including alloys, intermetallics and minerals. Commercial, access by subscription. 139058 entries (Dec 1, 2010). American Mineralogist Crystal Structure Database (AMCSD) (http://rruff.geo.arizona.edu/AMS/amcsd.php): Minerals. Free access (financed by NSF). 3156 different minerals (many more entries, as a mineral may appear at different pressures and temperatures). Reciprocal Net (http://www.reciprocalnet.org/): Small but well choosen set of quite common molecules and materials. Data from CSD and other sources. Free access (financed by NSF). Protein Data Bank (PDB) (http://www.rcsb.org/pdb/): Structures of large biological molecules, including proteins and nucleic acids. Free access (international support). 78992 structures (Jan 31, 2012). Indispensable portal for anyone working on biomolecules. Crystallography Open Database (COD) (http://www.crystallography.net/): Voluntary effort to provide an open alternative to CSD and ICSD. Absolutely free access. 158240 entries (feb 2, 2012) and growing fast. A sister PCOD database specializes on theoretically predicted structures (>1000000 in Nov 2009). Structural Classification of Proteins (SCOP) (http://scop.mrc-lmb.cam.ac.uk/scop/): Further analysis of the proteins contained in PDB: folds, superfamilies, evolutionary relationship, etc. Free access. Nucleic Acids Data Bank (NADB) (http://ndbserver.rutgers.edu/): Similar to PDB, but specialized on oligonucleotides. Free access (international support). 3745 structures (2008-01-17). V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 25 / 86
textbooks Crystal Data Bases II Mineralogy Database (http://webmineral.com/): Good database on minerals and gems maintained by commercial dealers. Free access. Crystal Lattice Structures (http://cst-www.nrl.navy.mil/lattice/): Very good description of the common crystal lattice structures of the elements and simple compounds. Free access. Powder diffraction file (PDF) (http://www.icdd.com/): Largest DB on single phase powder diffraction pattern. Widely used to identify compounds based on their fingerprint spectra. Commercial. Database of Macromolecular Movements (http://molmovdb.mbb.yale.edu/molmovdb/): Analysis and prediction of the dynamical behaviour of macromolecules. Movies, morphings, etc. Free access. Do you know any other good structures database? Please, e-mail me the address and details (mailto:[email protected]) V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 26 / 86
textbooks Quantum Espresso: pw+ps GNU code opEn Source Package for Research in Electronic Structure, Simulation, and Optimization PWSCF, CP, PHONON, FPMD, Wannier, ... Suite of codes for DFT electronic structure calculations and materials modeling. Based on the use of plane waves and pseudopotentials (both, norm-conserving and ultrasoft). http://www.quantum-espresso.org/ Installation from source is simple: 1 cd 2 mkdir -p src/qe/ 3 cd src/qe/ 4 wget http://qe-forge.org/frs/download.php/211/espresso-5.0.tar.gz 5 cd espresso-5.0 6 ./configure # finds compilers , l i b r a r i e s , and creates "make . sys " 7 ./make all # creates executables in ~/ src / qe / espresso −5.0/ bin / 8 cat << EOF >> ~/.bashrc 9 export QE_HOME=~/pkgs/qe/espresso-5.0/ 10 export PATH=${PATH}:${QE_HOME}bin: 11 EOF 12 source ~/.bashrc V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 28 / 86
textbooks Get run examples from /apps/zcam2012/practicas-qe/ The file qe_si.tgz contains inputs, pseudopotentials, and bash scripts to perform the calculations: 1 # Unpack the tgz f i l e in a d i r e c t o r y . . . 2 mkdir si 3 cd si 4 cp -p /apps/zcam2012/practicas_qe/qe_si.tgz 5 tar zxvf qe_si.tgz 6 # . . . t h i s creates a number of d i r e c t o r i e s 7 # 8 # bands dos energy ps pseudo rho tmp 9 # 10 # Each d i r e c t o r y contains a ∗∗∗ r u n i t . sh∗∗∗ s c r i p t f i l e . Check and 11 # modify them to s u i t your purposes . 12 # 13 # Notice the d i r e c t o r i e s : 14 # ( pseudo ) I t contains the pseudopotentials that w i l l be used in the 15 # t u t o r i a l . 16 # ( tmp ) I t w l l be used as scratch d i r e c t o r y . Clean t h i s d i r e c t o r y once 17 # you f i n i s h completely your work . V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 30 / 86
textbooks pwSCF: Input scheme (pw.x < input > output) I The input file for pwSCF is structured into mandatory and optional "&NAMELISTS" and "INPUT_CARDS". Each namelist contains a number of control variables, most of them with appropriate default values, but a few are essential and must always be specified. The complete set of variables is described in the "INPUT_PW.txt" file. &CONTROL: declare addresses and names for pseudopotentials, and other datafiles, select the amount of I/O (input/output), etc. Important variables: calculation ( task to be performed: ’scf’, ’nscf’, ’bands’, ’relax’, ’md’, ’vc-relax’, ’vc-md’). &SYSTEM: describe the system to be calculated. Enforced variables: ibrav (0–14, Bravais- lattice index). celldm (cell parameters required by the crystalline system); nat (number of atoms in the unit cell); ntyp (number of atomic types); ecutwfc (kinetic energy cutoff). &ELECTRONS: control the SCF process and the algorithm to be used. The most important variable is diagonalization, the main options being Davidson iterative (’david’) and conjugate gradient like (’cg’). &IONS: needed when atoms move (structural relaxation and molecular dynamics runs) and ignored otherwise. &CELL: needed when the cell moves (structural relaxation and variable sell MD runs) and ignored otherwise. &PHONON: prepare the input for a PHONON task. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 31 / 86
textbooks Each input card, on the other hand, corresponds to some vector or matrix. ATOMIC_SPECIES: <atom> <mass> <pseudopotential> for every different atomic species. ATOMIC_POSITIONS: <atom> <x> <y> <z> for every different atomic position. K_POINTS: <number-of-k-points> <x> <y> <z> <multiplicity> The k-points can correspond to the integration of iBZ1 or to special points used for depicting band structure. CELL_PARAMETERS: OCCUPATIONS: CLIMBING_IMAGES: special data for the NEB (nudge elastic band) algorithm. CONSTRAINTS: V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 32 / 86
textbooks pwSCF: Control of the input structure There are several methods to introduce the crystal structure in pwscf. They are controlled by ibrav, celldm, nat, and the CELL_PARAMETERS input card. ibrav 0 1 2 3 4 5 cell free cP (sc) cF (fcc) cI (bcc) hP hR(1) celldm (1) a (1) a (1) a (1) a (1) a (1) a (3) c/a (4) C = cos α v1 a(1, 0, 0) a(−1, 0, 1)/2 a(1, 1, 1)/2 a(1, 0, 0) a(tx, −ty, tz) v2 a(0, 1, 0) a(0, 1, 1)/2 a(−1, 1, 1)/2 a(−1/2, √ 3/2, 0) a(0, 2ty, tz) v3 a(0, 0, 1) a(−1, 1, 0)/2 a(−1, −1, 1)/2 a(0, 0, c/a) a(−tx, −ty, tz) • Cell: shows the crystalline system ([c]ubic, [t]etragonal, [o]rthorhombic, [h]exagonal or rhombohedric with hexagonal axes, [m]onoclinic, or [a]=triclininc) and the (P,I,F,A,B,C,R) centering. • hR: the threefold axis is either (1) c, or (2) the < 111 > direction. • hR: tx = (1 − C)/2; ty = (1 − C)/6; tz = (1 + 2C)/3; u = tz − 2 √ 2ty; v = tz + √ 2ty. The v1..v3 vectors, except for the a scale, form the R matrix that can be declared in the CELL_PARAMETERS input card. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 33 / 86
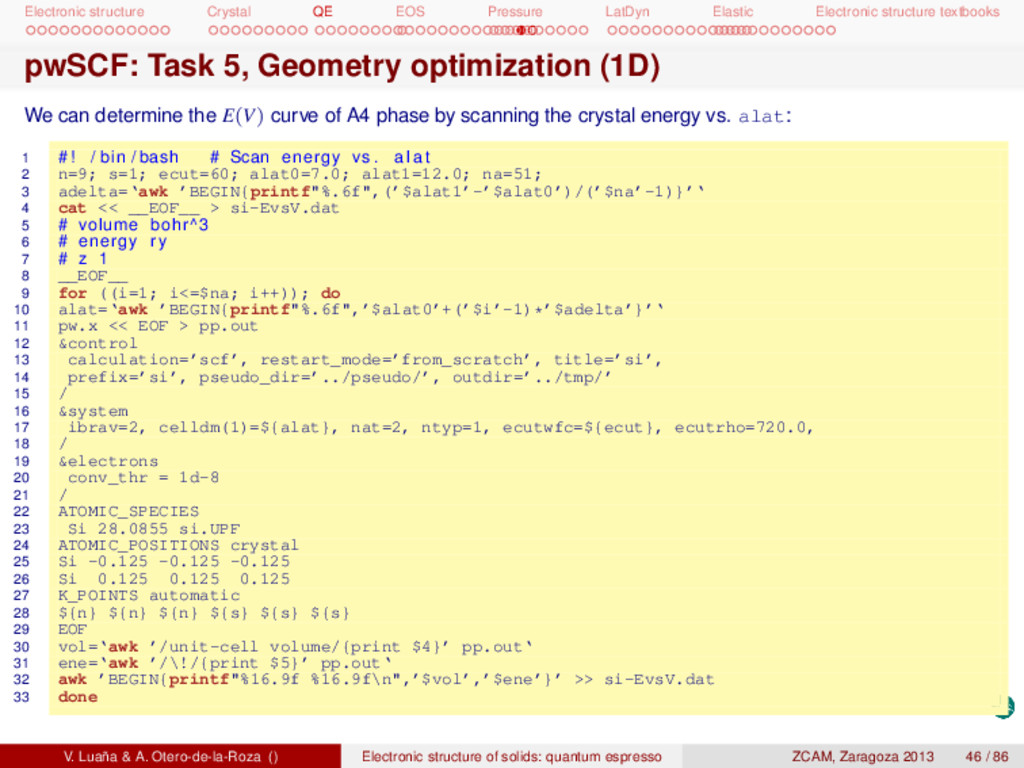
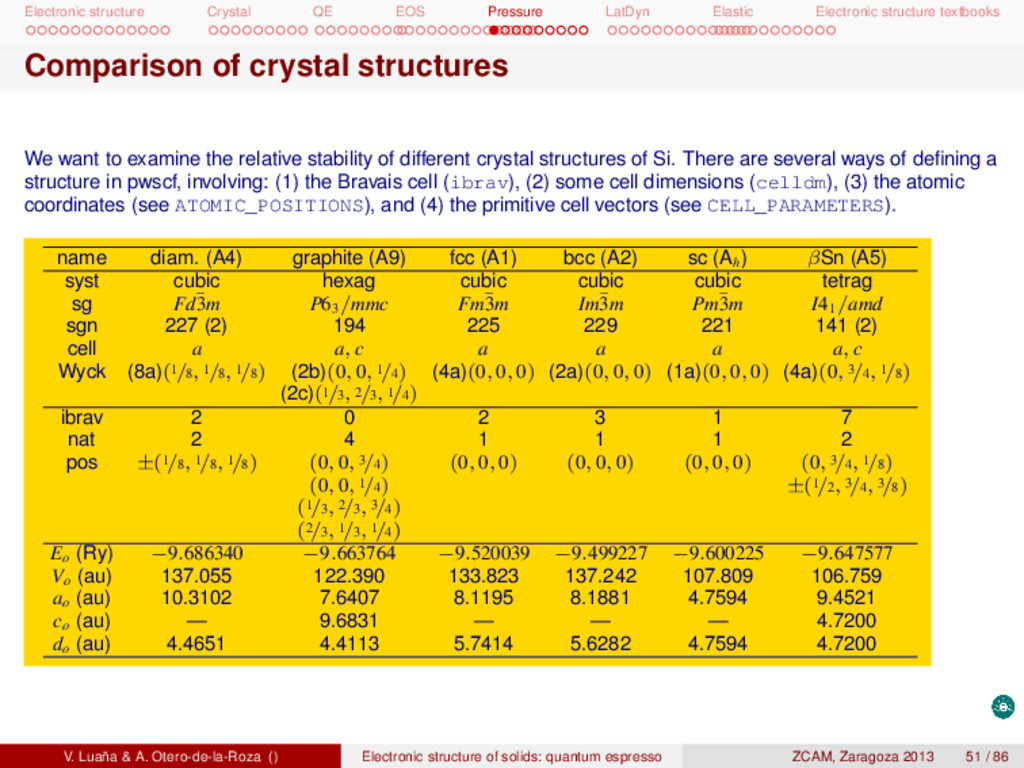
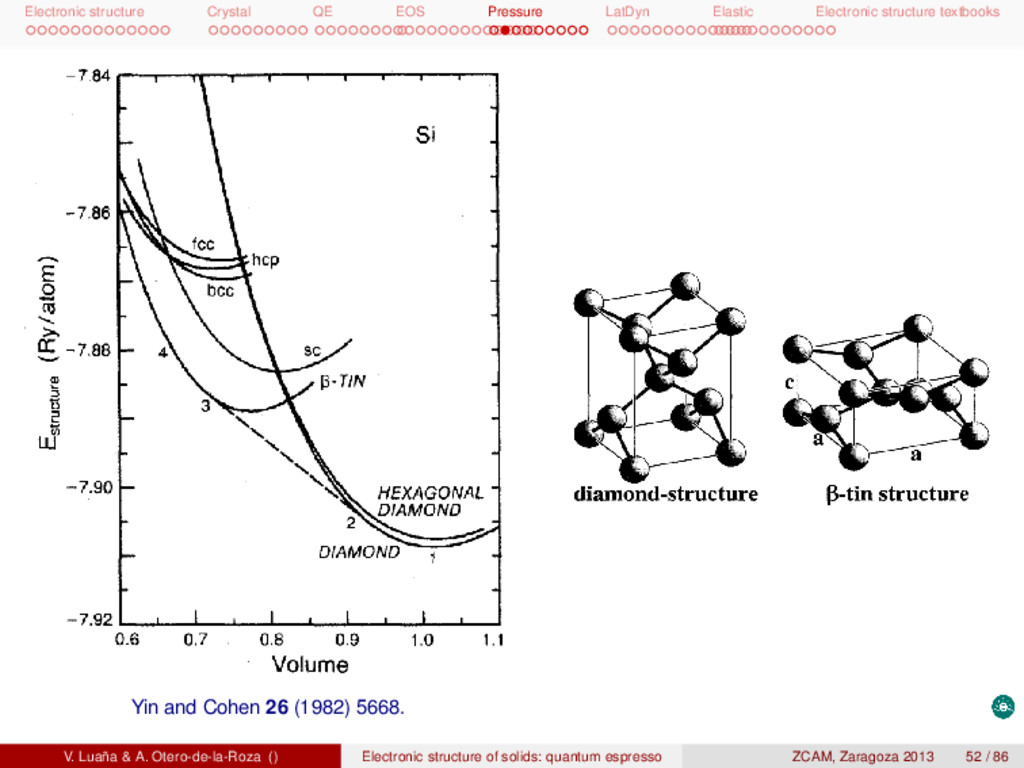
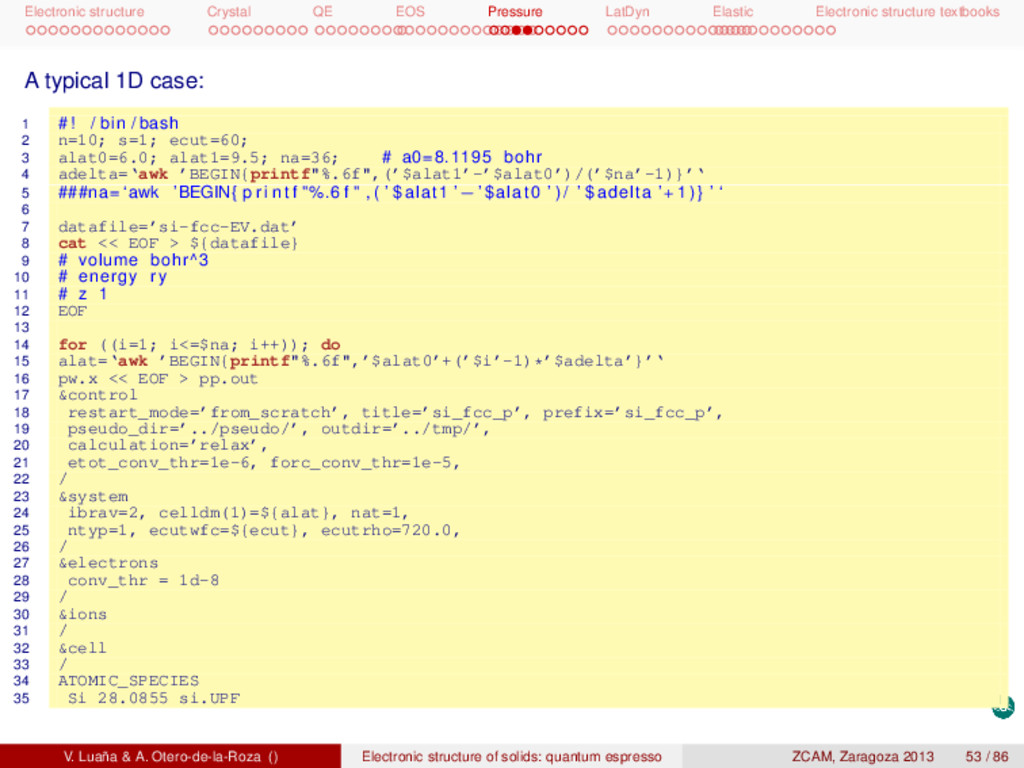
textbooks pwSCF: Task 1, energy at a single geometry I Remember to copy the tutorial files and install them in your account: 1 # Unpack the tgz f i l e in a d i r e c t o r y . . . 2 mkdir si 3 cd si 4 cp -p /apps/zcam2012/practicas_qe/qe-si.tgz 5 tar zxvf qe-si.tgz 6 # . . . t h i s creates a number of d i r e c t o r i e s 7 # 8 # bands dos energy ps pseudo rho tmp 9 # 10 # Each d i r e c t o r y contains a ∗∗∗ r u n i t . sh∗∗∗ s c r i p t f i l e . 11 # Calculation2 : 12 # (1) t o t a l energy of Si dimond phase at the experimental geometry 13 cd energy 14 pw.x < si.scf.in > si.scf.out 15 # Run the c a l c u l a t i o n several times exploring the e f f e c t of ecutfc and the 16 # size of the Monkhorst−Pack grid . Check ∗∗ r u n i t 2 . sh∗∗ and ∗∗ r u n i t 3 . sh∗∗. Other calculations are in the directories: (opt1) geometry optimization, (bands) band diagram, (dos) electronic density of states, (eos1) fit the E(V) data to an analytical equation of state, (ph-opt) equilibrium geometry for several phases of Si, (ph-eos) get the E(V) data for several phases, (cij) elastic constants, (phon) phonon calculation, .... V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 35 / 86
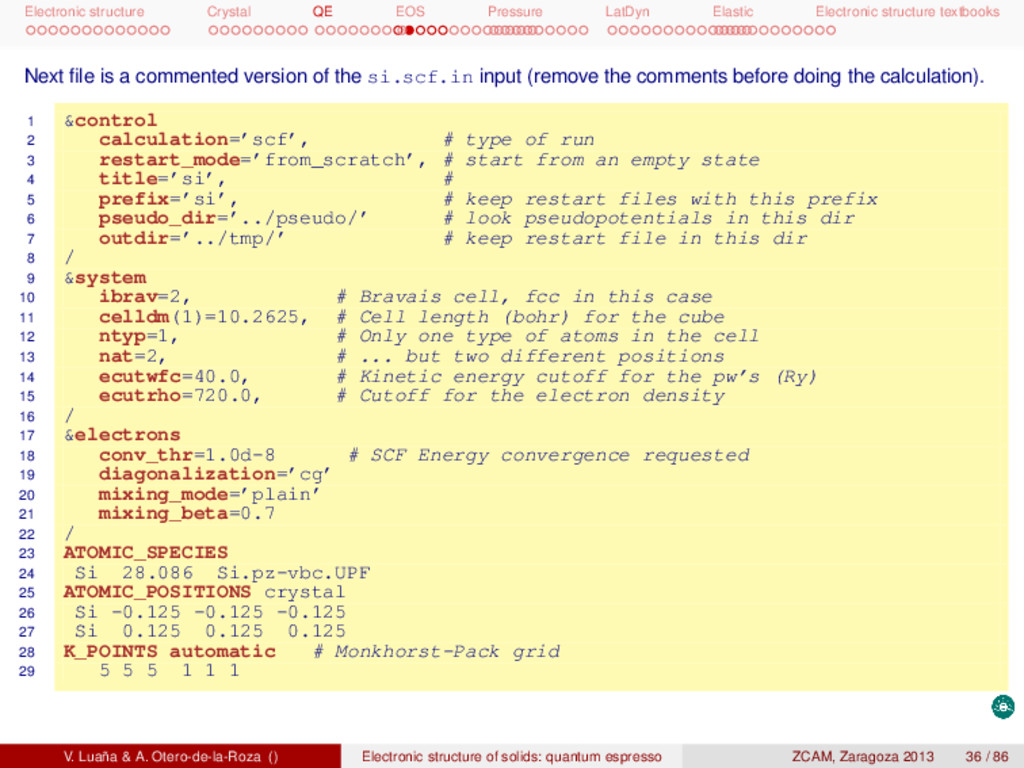
textbooks Next file is a commented version of the si.scf.in input (remove the comments before doing the calculation). 1 &control 2 calculation=’scf’, # type of run 3 restart_mode=’from_scratch’, # start from an empty state 4 title=’si’, # 5 prefix=’si’, # keep restart files with this prefix 6 pseudo_dir=’../pseudo/’ # look pseudopotentials in this dir 7 outdir=’../tmp/’ # keep restart file in this dir 8 / 9 &system 10 ibrav=2, # Bravais cell, fcc in this case 11 celldm(1)=10.2625, # Cell length (bohr) for the cube 12 ntyp=1, # Only one type of atoms in the cell 13 nat=2, # ... but two different positions 14 ecutwfc=40.0, # Kinetic energy cutoff for the pw’s (Ry) 15 ecutrho=720.0, # Cutoff for the electron density 16 / 17 &electrons 18 conv_thr=1.0d-8 # SCF Energy convergence requested 19 diagonalization=’cg’ 20 mixing_mode=’plain’ 21 mixing_beta=0.7 22 / 23 ATOMIC_SPECIES 24 Si 28.086 Si.pz-vbc.UPF 25 ATOMIC_POSITIONS crystal 26 Si -0.125 -0.125 -0.125 27 Si 0.125 0.125 0.125 28 K_POINTS automatic # Monkhorst-Pack grid 29 5 5 5 1 1 1 V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 36 / 86
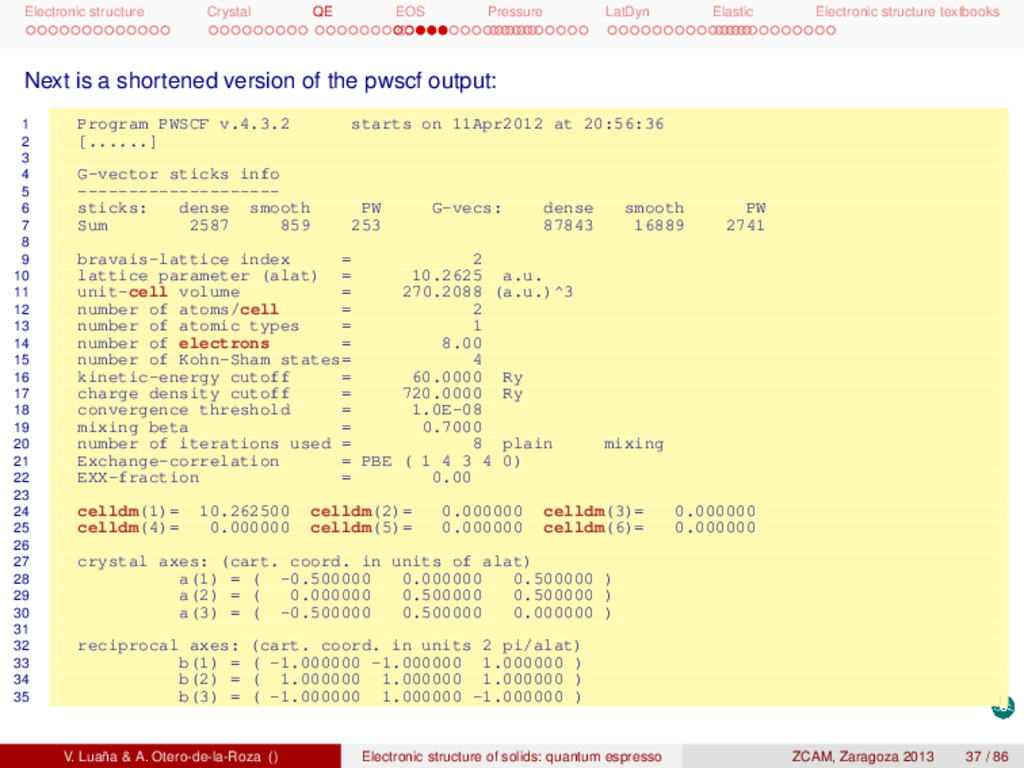
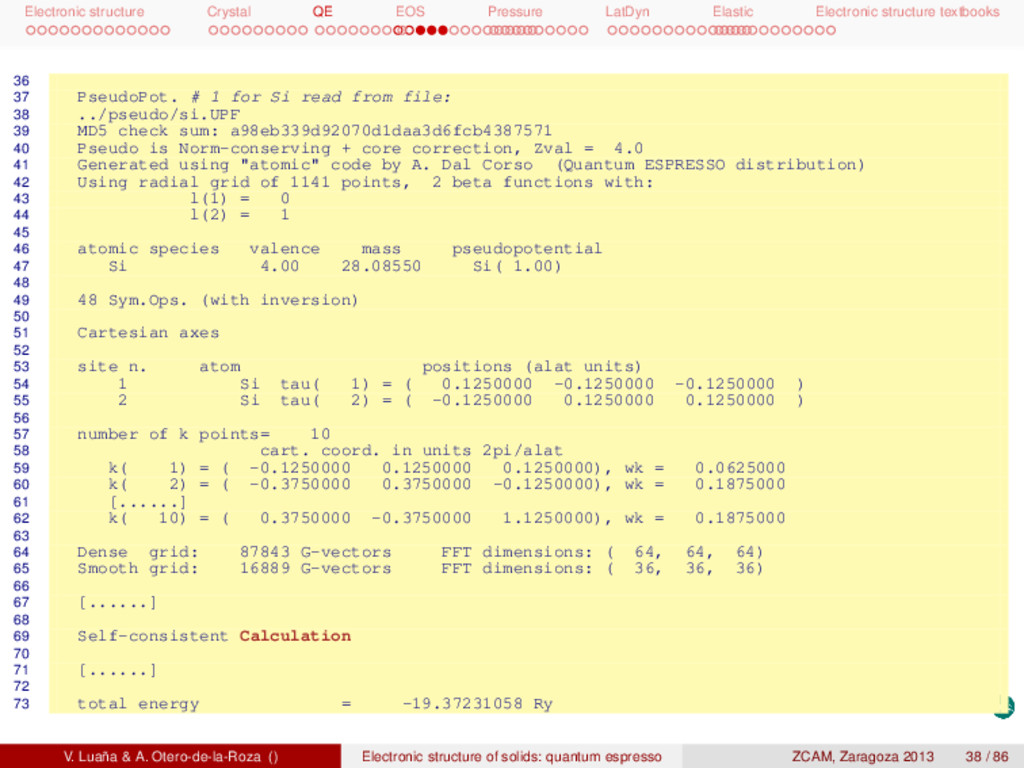
textbooks 74 Harris-Foulkes estimate = -19.37231059 Ry 75 estimated scf accuracy < 0.00000003 Ry 76 77 iteration # 6 ecut= 60.00 Ry beta=0.70 78 Davidson diagonalization with overlap 79 ethr = 3.45E-10, avg # of iterations = 2.6 80 81 total cpu time spent up to now is 4.0 secs 82 83 End of self-consistent calculation 84 85 k =-0.1250 0.1250 0.1250 ( 2112 PWs) bands (ev): 86 -5.5624 4.6045 5.9192 5.9192 87 [......] 88 k = 0.3750-0.3750 1.1250 ( 2125 PWs) bands (ev): 89 -2.8414 -0.4395 2.2179 4.3216 90 91 ! total energy = -19.37231059 Ry 92 Harris-Foulkes estimate = -19.37231059 Ry 93 estimated scf accuracy < 1.2E-09 Ry 94 95 The total energy is the sum of the following terms: 96 97 one-electron contribution = 5.02037625 Ry 98 hartree contribution = 1.09634466 Ry 99 xc contribution = -8.69219473 Ry 100 ewald contribution = -16.79683678 Ry 101 102 convergence has been achieved in 6 iterations 103 104 Writing output data file si.save 105 PWSCF : 3.99s CPU 4.08s WALL V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 39 / 86
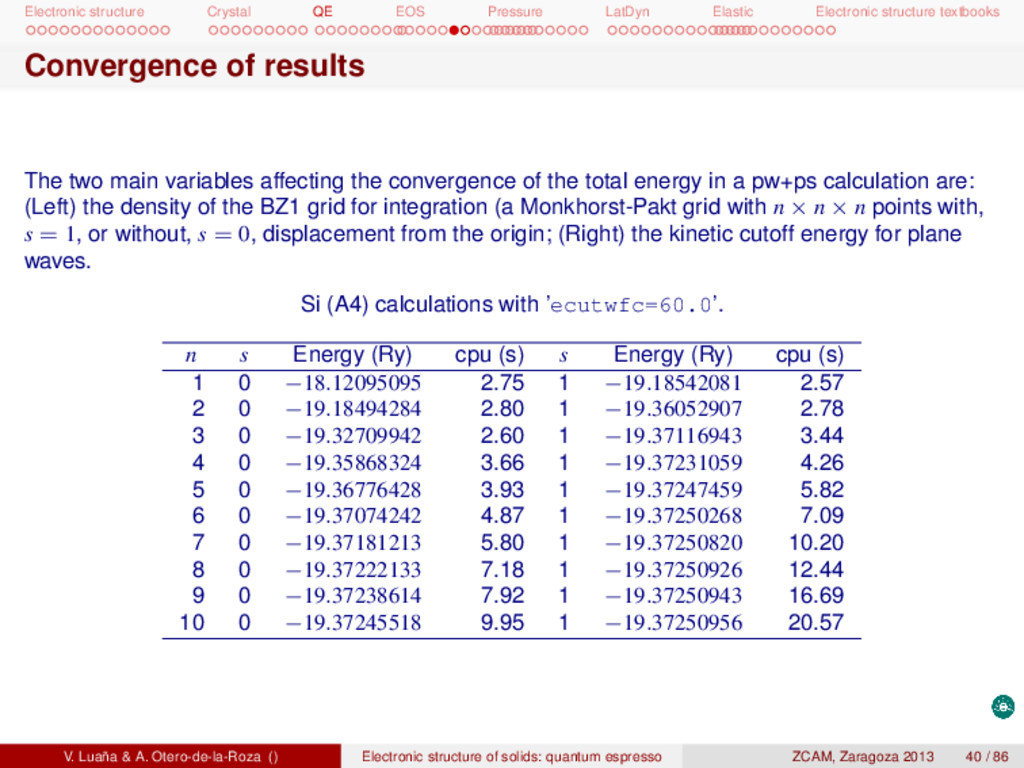
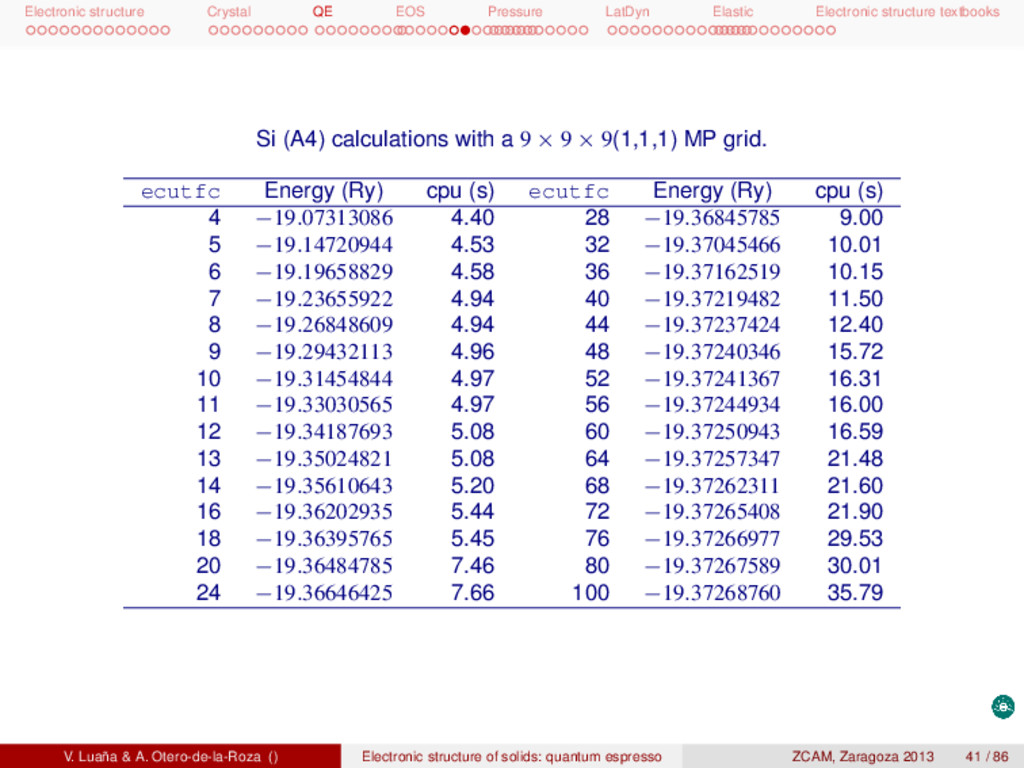
textbooks Convergence of results The two main variables affecting the convergence of the total energy in a pw+ps calculation are: (Left) the density of the BZ1 grid for integration (a Monkhorst-Pakt grid with n × n × n points with, s = 1, or without, s = 0, displacement from the origin; (Right) the kinetic cutoff energy for plane waves. Si (A4) calculations with ’ecutwfc=60.0’. n s Energy (Ry) cpu (s) s Energy (Ry) cpu (s) 1 0 −18.12095095 2.75 1 −19.18542081 2.57 2 0 −19.18494284 2.80 1 −19.36052907 2.78 3 0 −19.32709942 2.60 1 −19.37116943 3.44 4 0 −19.35868324 3.66 1 −19.37231059 4.26 5 0 −19.36776428 3.93 1 −19.37247459 5.82 6 0 −19.37074242 4.87 1 −19.37250268 7.09 7 0 −19.37181213 5.80 1 −19.37250820 10.20 8 0 −19.37222133 7.18 1 −19.37250926 12.44 9 0 −19.37238614 7.92 1 −19.37250943 16.69 10 0 −19.37245518 9.95 1 −19.37250956 20.57 V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 40 / 86
textbooks According to the current calculation, Si (A4 or diamond phase, a = 10.2625 Å) is a semiconductor, with an indirect bandgap of 0.6 eV (expt. value: 1.12 eV at 300 K). Notice that a good calculation of the band gap in a semiconductor is still one of the big unsolved problems of ab initio solid structure calculations. -15 -10 -5 0 5 10 L Γ X M Γ ε (eV) k-points V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 43 / 86
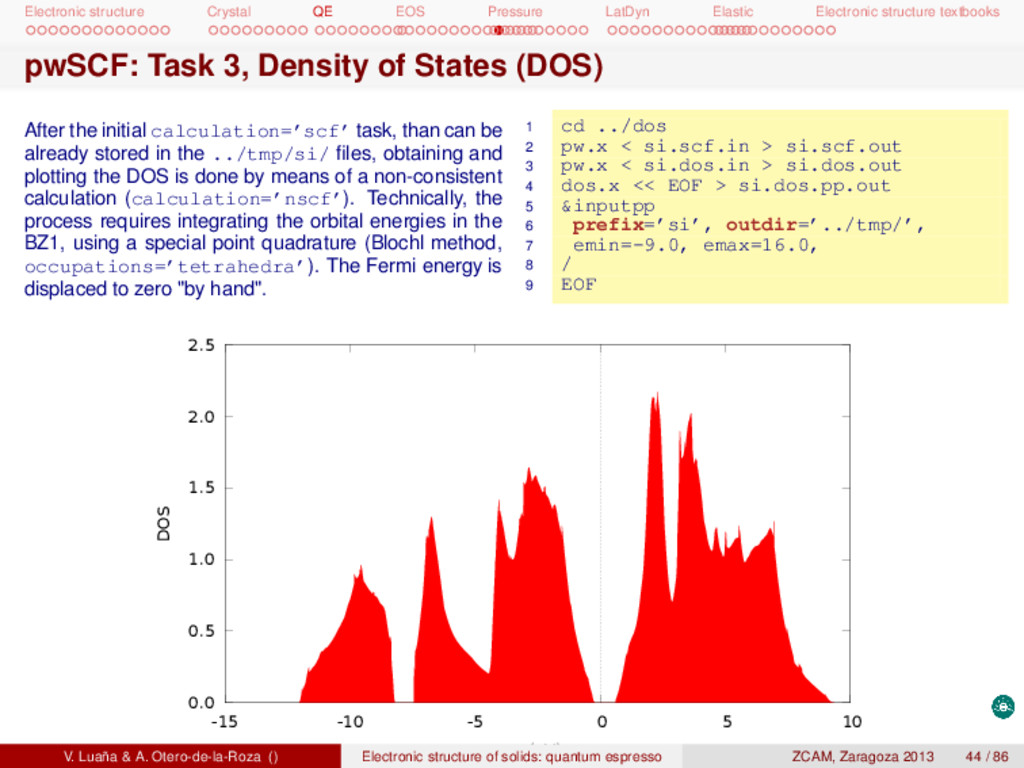
textbooks pwSCF: Task 3, Density of States (DOS) After the initial calculation=’scf’ task, than can be already stored in the ../tmp/si/ files, obtaining and plotting the DOS is done by means of a non-consistent calculation (calculation=’nscf’). Technically, the process requires integrating the orbital energies in the BZ1, using a special point quadrature (Blochl method, occupations=’tetrahedra’). The Fermi energy is displaced to zero "by hand". 1 cd ../dos 2 pw.x < si.scf.in > si.scf.out 3 pw.x < si.dos.in > si.dos.out 4 dos.x << EOF > si.dos.pp.out 5 &inputpp 6 prefix=’si’, outdir=’../tmp/’, 7 emin=-9.0, emax=16.0, 8 / 9 EOF 0.0 0.5 1.0 1.5 2.0 2.5 -15 -10 -5 0 5 10 DOS ε (eV) V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 44 / 86
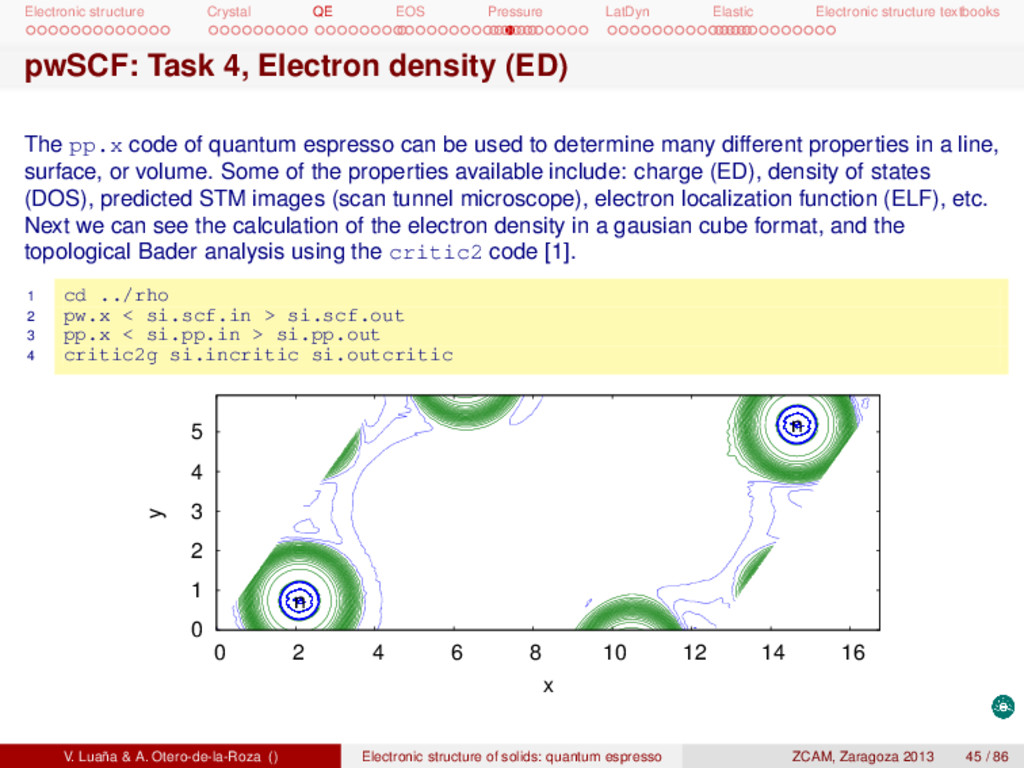
textbooks pwSCF: Task 4, Electron density (ED) The pp.x code of quantum espresso can be used to determine many different properties in a line, surface, or volume. Some of the properties available include: charge (ED), density of states (DOS), predicted STM images (scan tunnel microscope), electron localization function (ELF), etc. Next we can see the calculation of the electron density in a gausian cube format, and the topological Bader analysis using the critic2 code [1]. 1 cd ../rho 2 pw.x < si.scf.in > si.scf.out 3 pp.x < si.pp.in > si.pp.out 4 critic2g si.incritic si.outcritic 0 1 2 3 4 5 0 2 4 6 8 10 12 14 16 y x n n V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 45 / 86
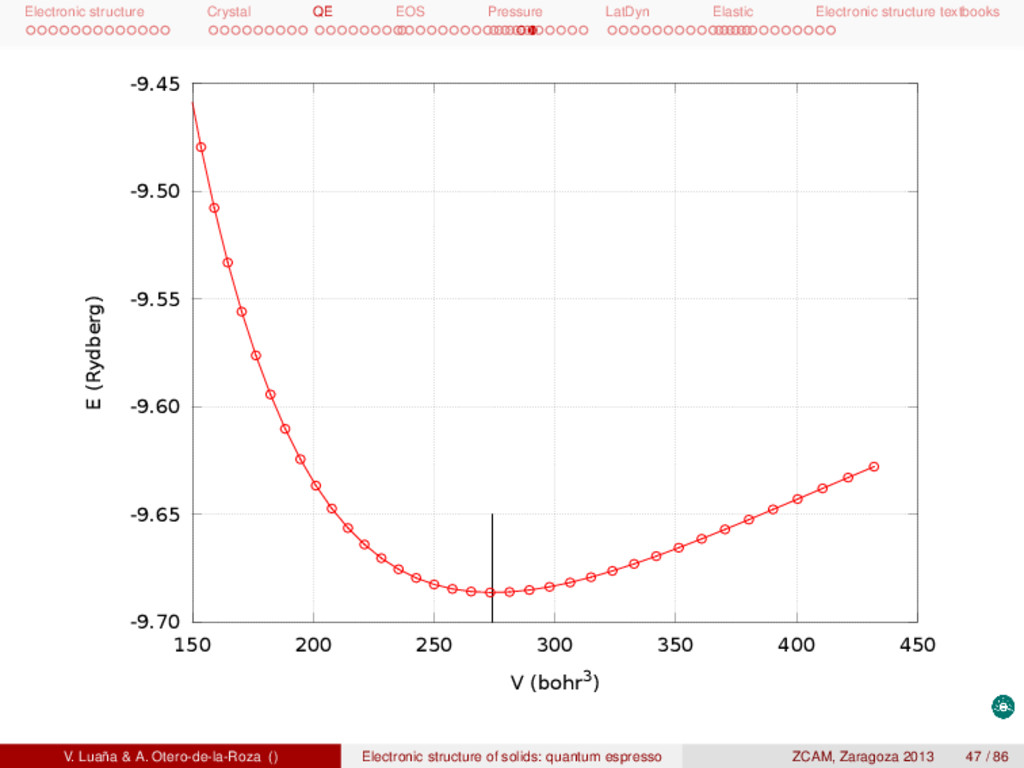
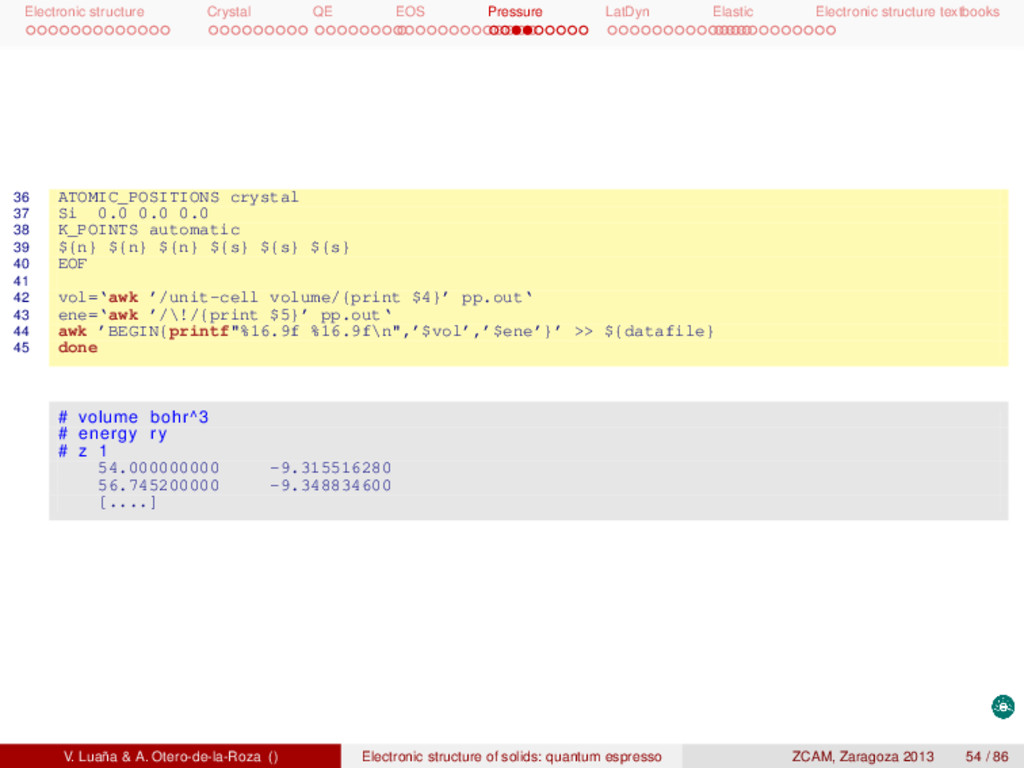
textbooks Pressure-volume equation of state (EOS) The asturfit package [2] can be used to fit the E(V) curve and determine the equilibrium properties and the pressure dependence of the mechanical properties of the crystal. These calculations are typically based on the static model, in which the entropy terms and the vibrational zero point energy are ignored. Under such conditions p = − ∂A ∂V NT ≈ − dE dV , G ≈ H ≈ E + pV, BT = −V ∂p ∂V NT ≈ V d2E dV2 . (16) The asturfit package [2] is an octave library of fitting routines but also implements three standalone tasks: (1) the recommended fitting procedure (asturfit, technically a linear fitting to an average of Birch-Murnaghan EOS of different degrees); (2) fit many different EOS to a E(V) dataset (allfits); and (3) analysis of possible noise and anomalies in the dataset (checknoise). A typical run is 1 asturfit << EOF > fit.out 2 # volume bohr^3 # Volume and 3 # energy ry # energy are given 4 # z 1 # per atom 5 85.750000000 -17.067433730 6 89.477700000 -17.278803700 7 [......] 8 EOF V0 (bohr3) E0 (Rydberg) B0 (GPa) B 0 B 0 (GPa−1) <BM> 274.1441(49) −9.686337(1) 88.6899(99) 4.2847(23) −0.077179(26) exp. 270.26 97.6 [3] V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 49 / 86
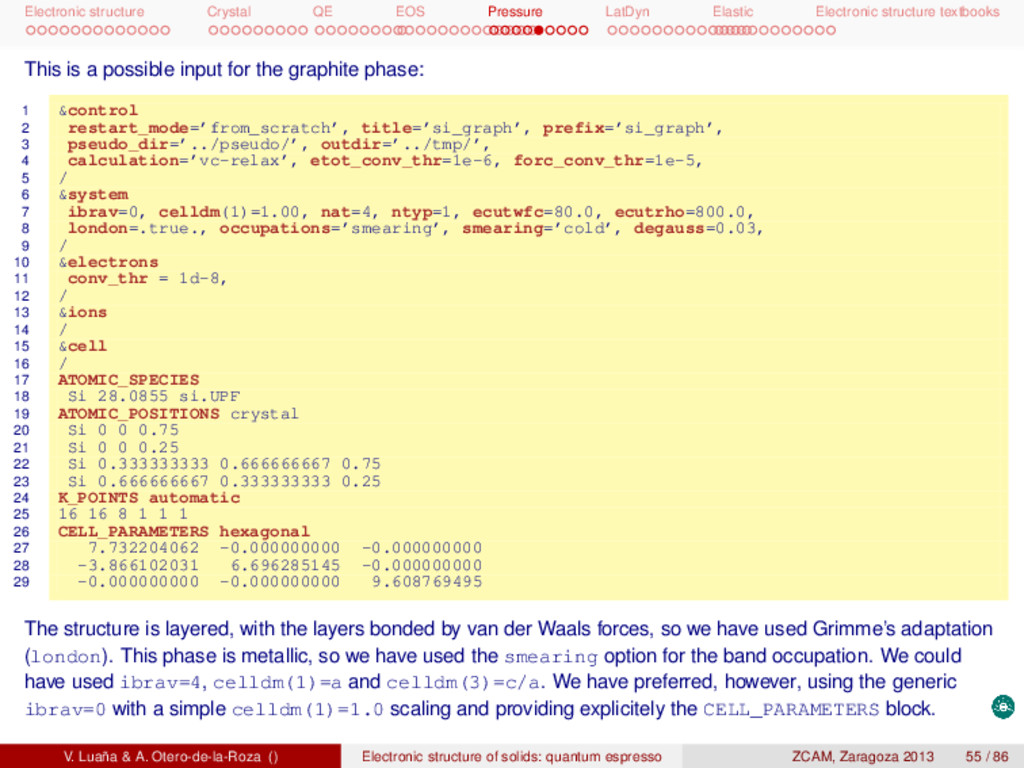
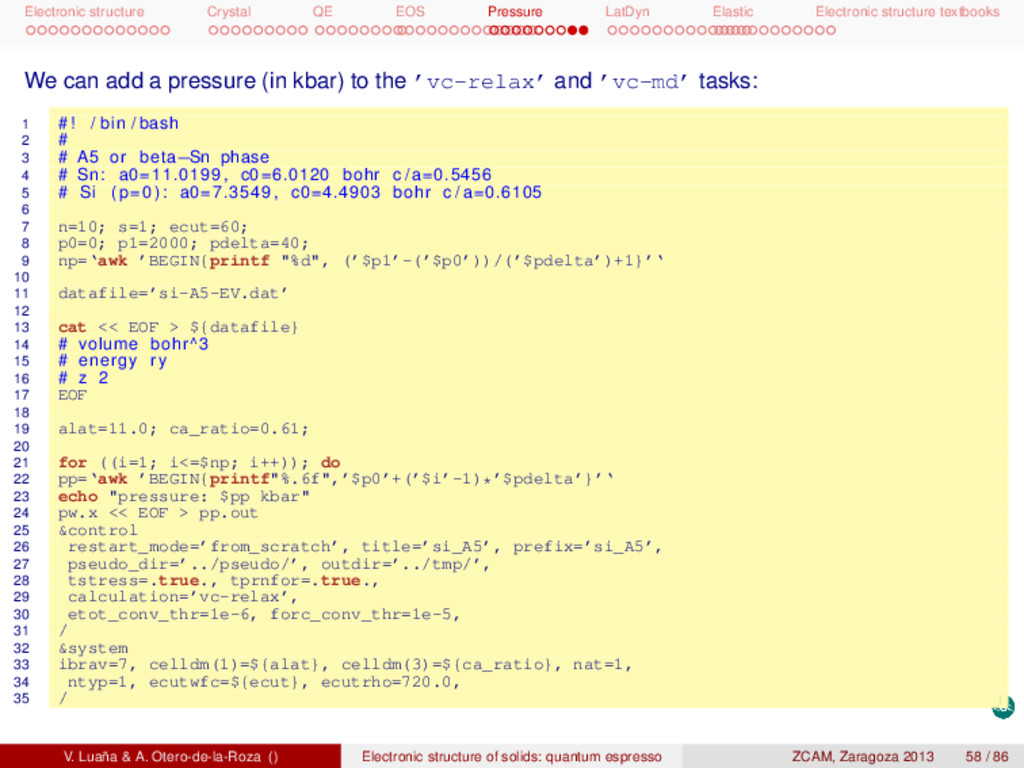
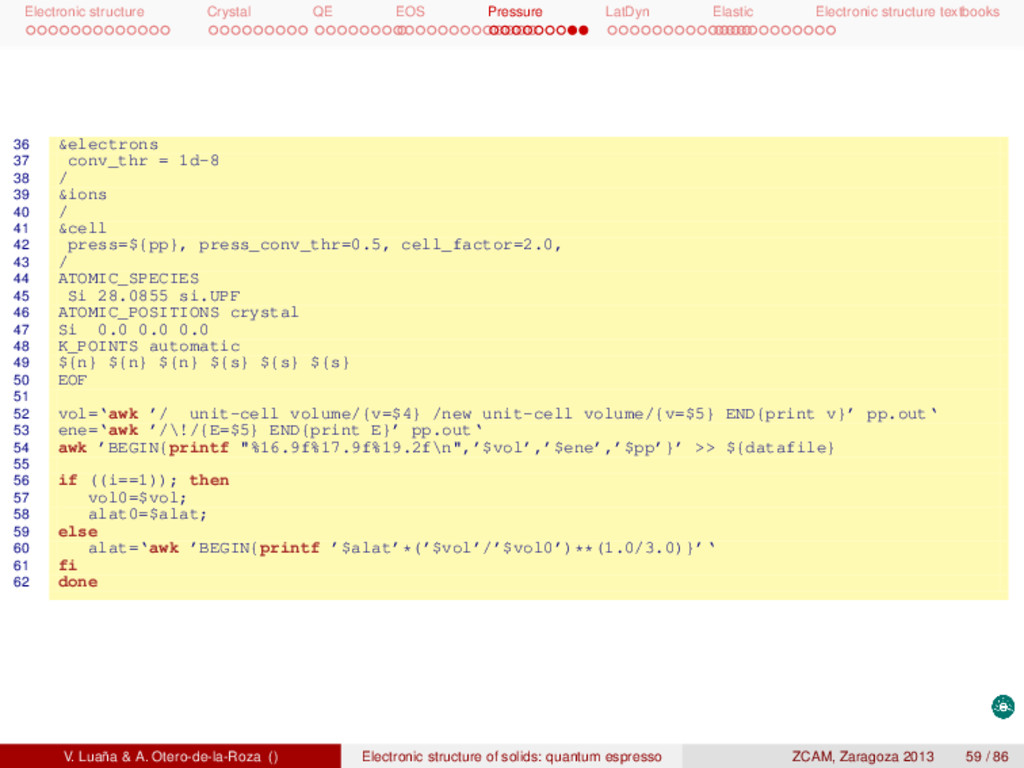
textbooks This is a possible input for the graphite phase: 1 &control 2 restart_mode=’from_scratch’, title=’si_graph’, prefix=’si_graph’, 3 pseudo_dir=’../pseudo/’, outdir=’../tmp/’, 4 calculation=’vc-relax’, etot_conv_thr=1e-6, forc_conv_thr=1e-5, 5 / 6 &system 7 ibrav=0, celldm(1)=1.00, nat=4, ntyp=1, ecutwfc=80.0, ecutrho=800.0, 8 london=.true., occupations=’smearing’, smearing=’cold’, degauss=0.03, 9 / 10 &electrons 11 conv_thr = 1d-8, 12 / 13 &ions 14 / 15 &cell 16 / 17 ATOMIC_SPECIES 18 Si 28.0855 si.UPF 19 ATOMIC_POSITIONS crystal 20 Si 0 0 0.75 21 Si 0 0 0.25 22 Si 0.333333333 0.666666667 0.75 23 Si 0.666666667 0.333333333 0.25 24 K_POINTS automatic 25 16 16 8 1 1 1 26 CELL_PARAMETERS hexagonal 27 7.732204062 -0.000000000 -0.000000000 28 -3.866102031 6.696285145 -0.000000000 29 -0.000000000 -0.000000000 9.608769495 The structure is layered, with the layers bonded by van der Waals forces, so we have used Grimme’s adaptation (london). This phase is metallic, so we have used the smearing option for the band occupation. We could have used ibrav=4, celldm(1)=a and celldm(3)=c/a. We have preferred, however, using the generic ibrav=0 with a simple celldm(1)=1.0 scaling and providing explicitely the CELL_PARAMETERS block. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 55 / 86
textbooks Statistical thermodynamics of vibrations I Let the crystal be formed by N = rM atoms vibrating in 3D, where r is the number of atoms in each of the M unit cells. All the 3N degrees of freedom of movement correspond to vibrations, as the formally infinite crystal has no translations nor rotations. Asuming harmonicity and independence of modes, each one of the 3N vibrations is associated to a normal mode of frequency ωi = 2πνi = 2π ki/µi , where ki and µi are the effective force constant and mass, respectively. The vibrational state of the i-th mode is described by a quantum number vi ∈ {0, 1, 2, 3, ...} and has an energy i = (vi + 1/2) ωi . The vibrational state and energy of the whole crystal is described by |v = |v1 , v2 , ..., v3N and Ev = 3N i=1 (vi + 1/2) ωi. (17) No restrictions exist on the collective vibrational quantum numbers, as vibrations behave as bosons with null spin. In fact, the ground state corresponds to v = 0, all vibrational normal modes inactive, and the vibrational energy for this ground state is E0 = 3N i ωi/2. Using a classical, Boltzmann formulation for the canonical ensamble, the vibrational partition function is Q(3N, V, T) = v e−Ev/kT = ∞ v1=0 ... ∞ v3N =0 exp − 3N i=1 (vi + 1/2) ωi/kT = 3N i=1 ∞ vi=0 exp − (vi + 1/2) ωi kT = 3N i=1 e− ωi/2kT ∞ vi=0 e− ωi/kT vi = 3N i=1 e− ωi/2kT 1 − e− ωi/kT , (18) V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 61 / 86
textbooks Statistical thermodynamics of vibrations II where k is the Boltzmann constant, T the absolute temperature, and we have used the summation formula S = 1/(1 − r) for an infinite series, 1 + r + r2 + r3 + ..., of a convergent r = exp(− ω/kT) < 1 geometric progression. The thermodynamic potential directly related to the canonical partition function is the Helmholtz function: A(3N, V, T) = −kT ln Q(3N, V, T) = 3N i=1 ωi 2 + kT ln 1 − e− ωi/kT , (19) and all the thermodynamic properties can be derived from here. For instance: E = − ∂ ln Q ∂β N,V , p = − ∂A ∂V N,T , S = k ln Q + E/T, ... (20) where β = 1/kT. In a macroscopic crystal 3N is huge (of the order on the Avogadro number), and the summation over normal modes can be converted to an integration: A(3N, V, T) = ∞ 0 dωg(ω) ωi 2 + kT ln 1 − e− ωi/kT , (21) where g(ω) is the vibrational density of states (DOS), normalized as ∞ 0 g(ω)dω = 3N. (22) The DOS represents the degeneracy of normal modes. Knowledge of DOS is all that is required to determine the vibrational contribution to the thermodinamical properties of the crystal under the harmonic approach. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 62 / 86
textbooks Einstein, Debye and QH Debye models I The Einstein model [Ann. Physik 22 (1907) 180] Used to explain the main aspects of the heat capacity of solids. All crystal atoms vibrate with the same frequency: g(ω)dω = 3Nδ(ω − ωE)dω ⇒ Cv = 3Nk ωE 2kT 2 1 sinh2( ωE/2kT) (23) Increases from Cv(T → 0) = 0 to the Dulong-Petit limit Cv(T → ∞) = 3Nk. Decays exponentially as T → 0. No explanation for thermal expansion. The Debye model [Ann. Physik 39 (1912) 789] The solid is modelled as an elastic continuous medium: g(ω)dω = 9N ω3 D ω2dω if ω ≤ ωD, 0 if ω > ωD, ⇒ Cv = 9Nk T θD 3 θD/T 0 x4exdx (ex − 1)2 (24) where ωD represents the highest frequency that could be propagated by the crystal, and θD = ωD/k is the Debye temperature. Cv(T → 0) ≈ (T/θD)3 and Cv(T → ∞) = 3Nk. No explanation for thermal expansion. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 63 / 86
textbooks Quasiharmonic Debye model [Blanco et al. CPC 158 (2004) 57] The Debye model is used, but the Debye frequency is related to the static bulk modulus, which is different for different volumes: B(V) = V ∂2A dV2 ≈ V ∂2E dV2 ⇒ θD(V) = k (6π2V1/2r)1/3f(σ) B m , (25) where m is the molecular mass per formula unit, σ is the Poisson ratio, and f(σ) = 3 2 2(1 + 2σ) 3(1 − 2σ) 3/2 + 1 + σ 3(1 − σ) 3/2 1/3 . (26) In an isotropic solid σ = λ/2(λ + µ), where λ and µ are the coefficients of Lamé. For a cubic solid: c11 ≈ λ + µ, c12 ≈ λ, and c44 ≈ µ, so σ ≈ c12 /(c11 + c12 ) ≈ c12 /2(c12 + c44 ) ≈ 1/4. The vibrational contribution to the free energy is then Avib(T, V) = rMkT 9θ 8T + 3 ln(1−e−θ/T ) − D3 (θ/T) , D3 (x) = 3 x3 x 0 t3dt et − 1 . (27) Thermal behaviour is inferred from E(V) data. Correct limits for Cv when T → 0 and T → ∞. Thermal expansion is explained for normal materials at low T. No intrinsic anharmonicity. Fusion? V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 64 / 86
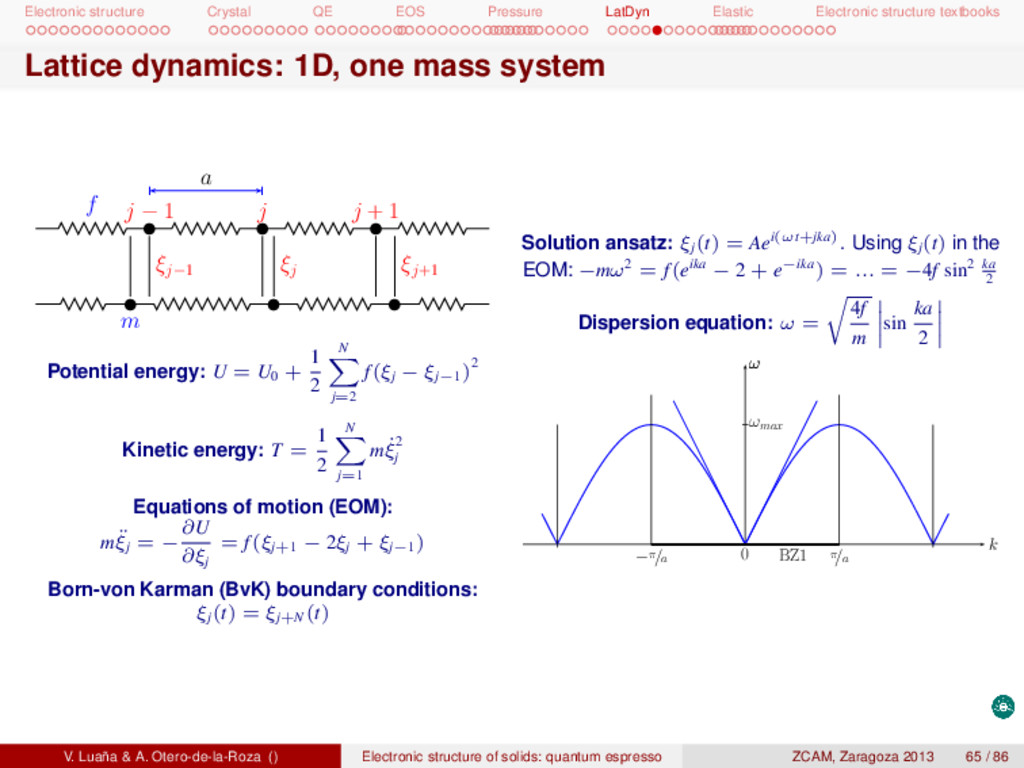
textbooks Example: 1D, two masses system I Hamiltonian: a f 2j − 2 2j − 1 2j 2j + 1 ξ2j−2 ξ2j−1 ξ2j ξ2j+1 m M H = T + U = 1 2 N j=1 (m ˙ ξ2 2j + M ˙ ξ2 2j−1 ) + U0 + 1 2 N j=1 f[(ξ2j − ξ2j−1 )2 + (ξ2j+1 − ξ2j)2] Equations of motion (EOM): m¨ ξ2j = f(ξ2j+1 +ξ2j−1 −2ξ2j), M ¨ ξ2j+1 = f(ξ2j+2 +ξ2j−2ξ2j+1 ). Periodic conditions: ξj+2N (t) = ξj(t). Solution ansatz: ξ2j(t) = Aei(ωt+2jka) and ξ2j+1 (t) = Bei(ωt+(2j+1)ka). Dynamical equation: 2f − mω2 −2f cos ka −2f cos ka 2f − Mω2 A B = 0, k ∈ [−π/2a, π/2a] Eigenvalues: ω4 − 2f µ ω2 + 4f2 sin2 ka mM = 0 ⇒ ω2 ± = f µ 1 ± 1 − 4nM sin2 ka (m + M)2 where µ = mM/(m + M) is the reduced mass of the A and B particles. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 66 / 86
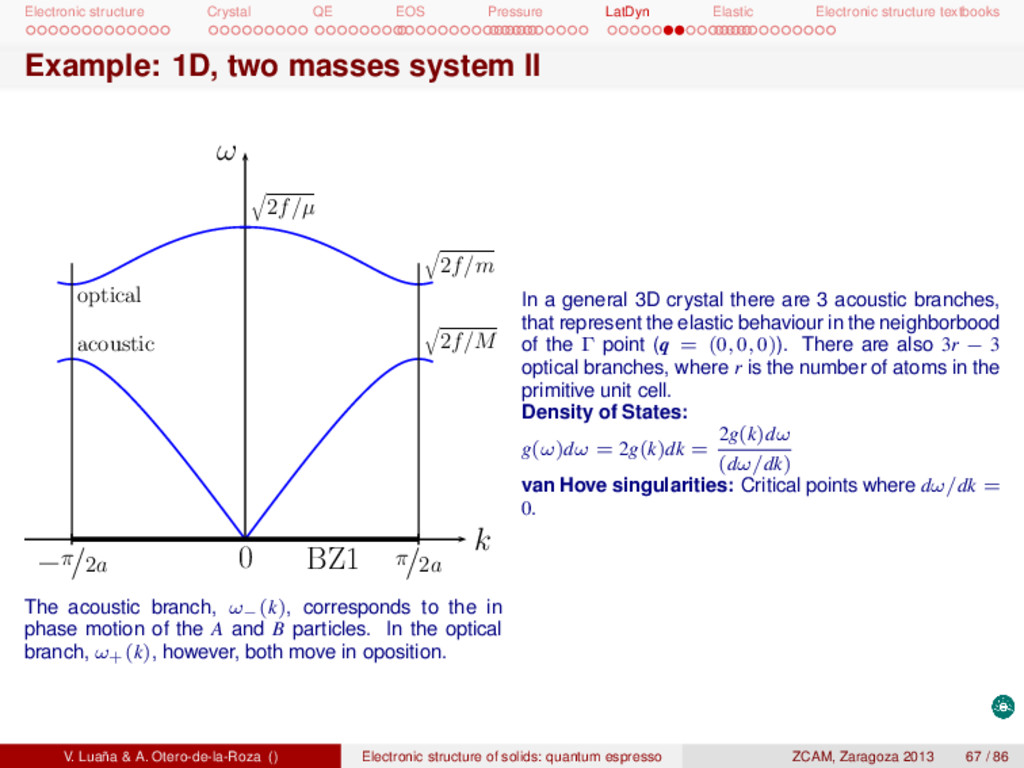
textbooks Example: 1D, two masses system II 0 BZ1 π/2a −π/2a 2f/µ 2f/m 2f/M k ω acoustic optical The acoustic branch, ω−(k), corresponds to the in phase motion of the A and B particles. In the optical branch, ω+(k), however, both move in oposition. In a general 3D crystal there are 3 acoustic branches, that represent the elastic behaviour in the neighborbood of the Γ point (q = (0, 0, 0)). There are also 3r − 3 optical branches, where r is the number of atoms in the primitive unit cell. Density of States: g(ω)dω = 2g(k)dk = 2g(k)dω (dω/dk) van Hove singularities: Critical points where dω/dk = 0. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 67 / 86
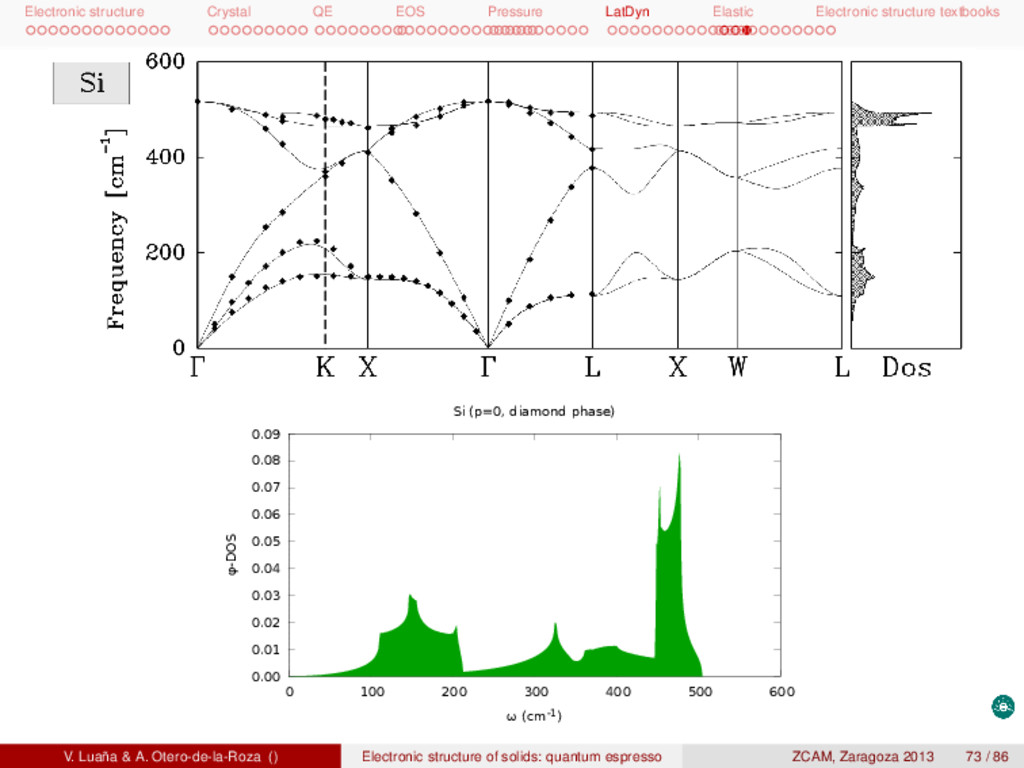
textbooks Lattice dynamics: general 3D crystal Dynamical equations (eigensystem) To be solved for q ∈ BZ1: κ β (˜ Dκα,κ β (q) − ω2 mq δκ,κ δα,β)γmq(κ β) = 0. (28) κ, κ : crystal primitive unit cells; α, β: atoms in a cell; q: reciprocal space vector; m: branch index. Dynamical matrix: Obtained using Density Functional Perturbation Theory (DFPT) or finite differences: ˜ Dκα,κ β (q) = 1 √ MκMκ cells b ∂2E ∂u0 κα ∂ub κ β eiqRb (29) E: energy of a unit cell (electronic and ionic); Rb: origin of the b cell; ua κα (t): instantaneous displacement (alpha: x, y, z) from the equilibrium position of the κ atom of cell a. Eigenvector displacement: ua κα (t) = 1 √ Mκ γmq(κα)ei(qRa−ωmqt). (30) V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 68 / 86
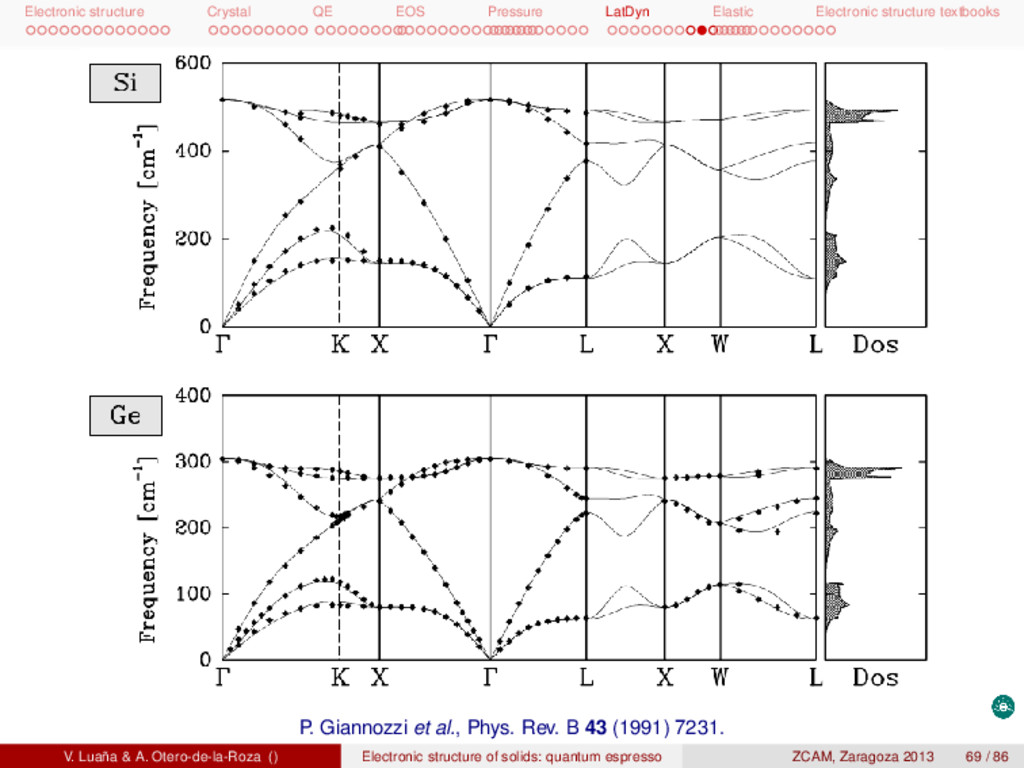
textbooks P. Giannozzi et al., Phys. Rev. B 43 (1991) 7231. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 69 / 86
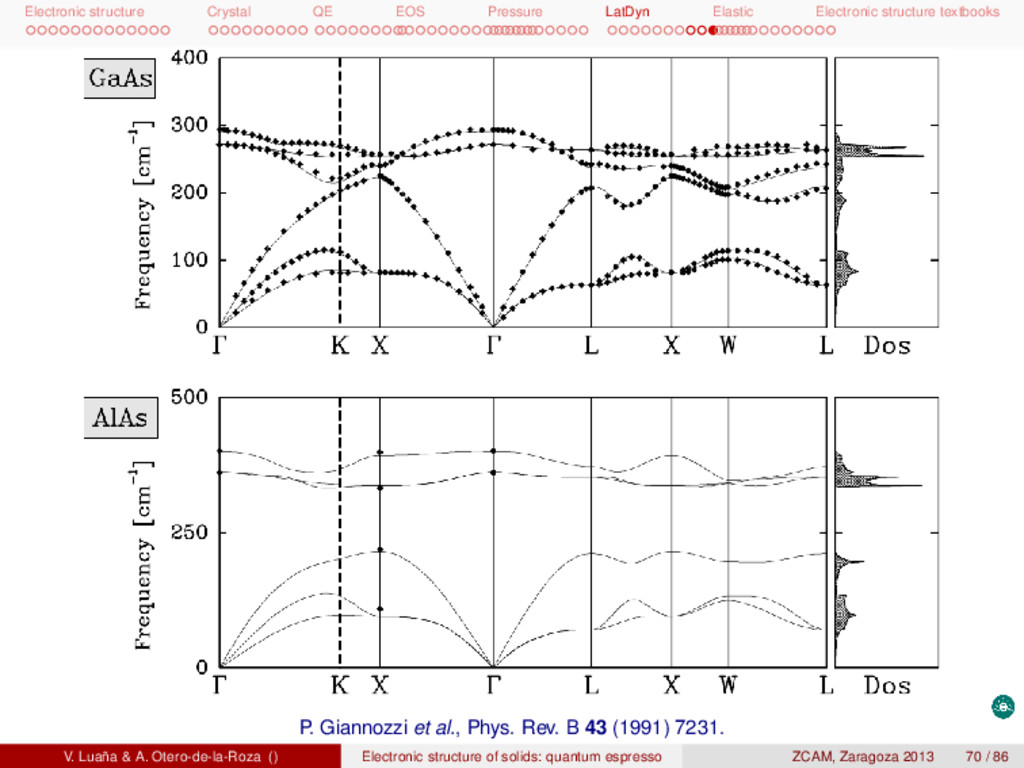
textbooks P. Giannozzi et al., Phys. Rev. B 43 (1991) 7231. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 70 / 86
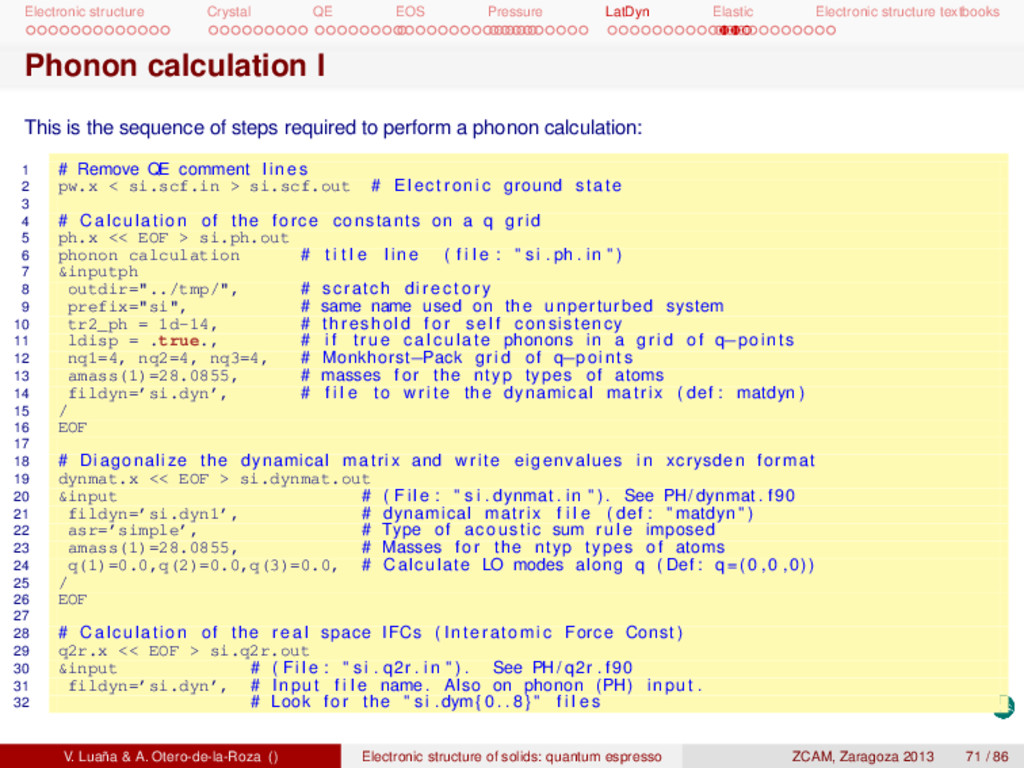
textbooks Phonon calculation I This is the sequence of steps required to perform a phonon calculation: 1 # Remove QE comment l i n e s 2 pw.x < si.scf.in > si.scf.out # Electronic ground state 3 4 # Calculation of the force constants on a q grid 5 ph.x << EOF > si.ph.out 6 phonon calculation # t i t l e l i n e ( f i l e : " s i . ph . in " ) 7 &inputph 8 outdir="../tmp/", # scratch d i r e c t o r y 9 prefix="si", # same name used on the unperturbed system 10 tr2_ph = 1d-14, # threshold f o r s e l f consistency 11 ldisp = .true., # i f true calculate phonons in a grid of q−points 12 nq1=4, nq2=4, nq3=4, # Monkhorst−Pack grid of q−points 13 amass(1)=28.0855, # masses f o r the ntyp types of atoms 14 fildyn=’si.dyn’, # f i l e to write the dynamical matrix ( def : matdyn ) 15 / 16 EOF 17 18 # Diagonalize the dynamical matrix and write eigenvalues in xcrysden format 19 dynmat.x << EOF > si.dynmat.out 20 &input # ( F i l e : " s i . dynmat . in " ) . See PH/ dynmat . f90 21 fildyn=’si.dyn1’, # dynamical matrix f i l e ( def : " matdyn " ) 22 asr=’simple’, # Type of acoustic sum rule imposed 23 amass(1)=28.0855, # Masses f o r the ntyp types of atoms 24 q(1)=0.0,q(2)=0.0,q(3)=0.0, # Calculate LO modes along q ( Def : q =(0 ,0 ,0)) 25 / 26 EOF 27 28 # Calculation of the real space IFCs ( Interatomic Force Const ) 29 q2r.x << EOF > si.q2r.out 30 &input # ( F i l e : " s i . q2r . in " ) . See PH/ q2r . f90 31 fildyn=’si.dyn’, # Input f i l e name. Also on phonon (PH) input . 32 # Look f o r the " s i .dym { 0 . . 8 } " f i l e s V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 71 / 86
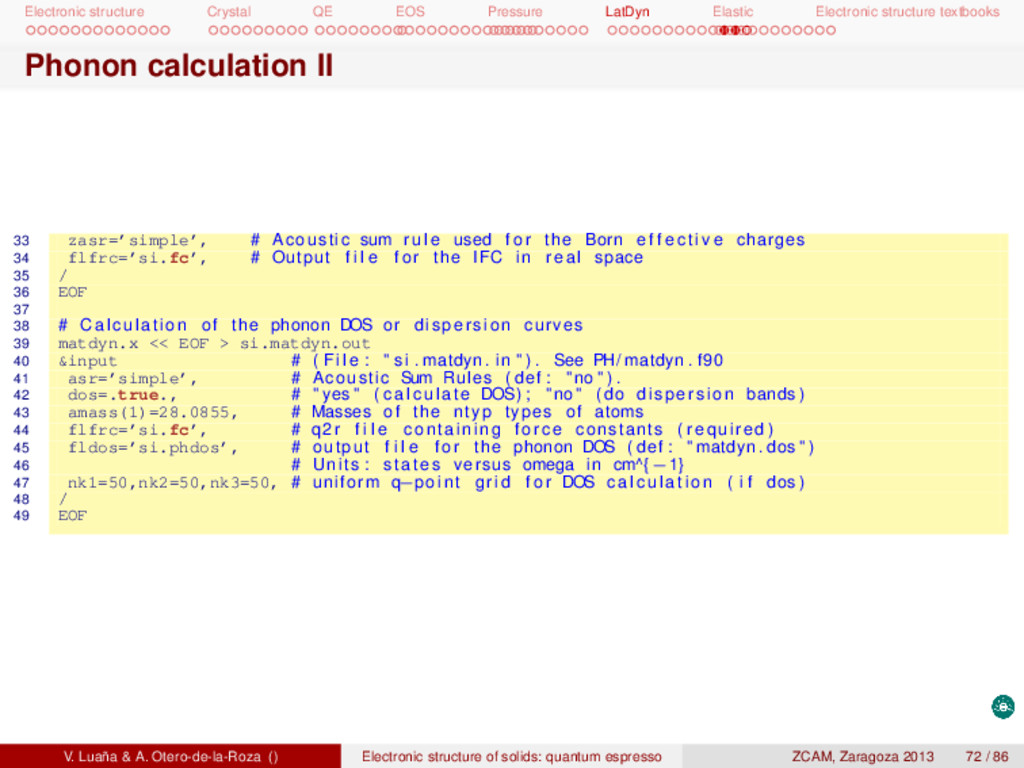
textbooks Phonon calculation II 33 zasr=’simple’, # Acoustic sum rule used f o r the Born e f f e c t i v e charges 34 flfrc=’si.fc’, # Output f i l e f o r the IFC in real space 35 / 36 EOF 37 38 # Calculation of the phonon DOS or dispersion curves 39 matdyn.x << EOF > si.matdyn.out 40 &input # ( F i l e : " s i . matdyn . in " ) . See PH/ matdyn . f90 41 asr=’simple’, # Acoustic Sum Rules ( def : " no " ) . 42 dos=.true., # " yes " ( calculate DOS) ; " no " ( do dispersion bands ) 43 amass(1)=28.0855, # Masses of the ntyp types of atoms 44 flfrc=’si.fc’, # q2r f i l e containing force constants ( required ) 45 fldos=’si.phdos’, # output f i l e f o r the phonon DOS ( def : " matdyn . dos " ) 46 # Units : states versus omega in cm^{−1} 47 nk1=50,nk2=50,nk3=50, # uniform q−point grid f o r DOS c a l c u l a t i o n ( i f dos ) 48 / 49 EOF V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 72 / 86
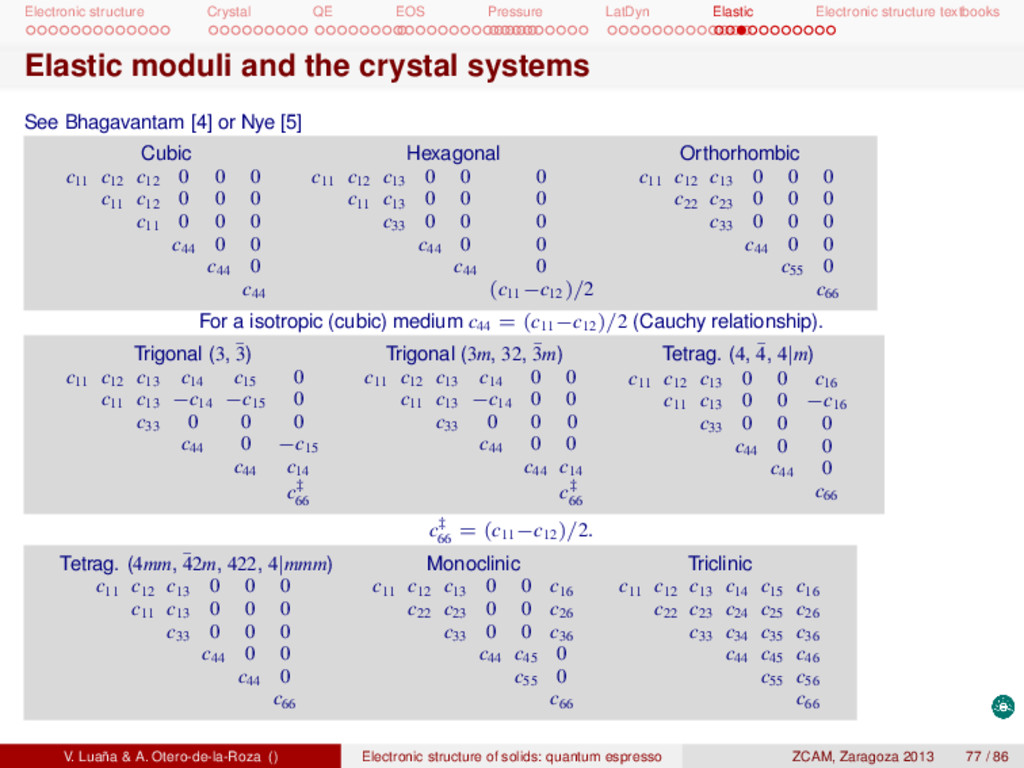
textbooks Catti’s method I In the method pioneered by M. Catti [6, 7] deformations involve simultaneous activation of several components of such that the crystal symmetry is maximally conserved. For instance, the deformation T[η, 0, 0, 0, 0, 0] in a cubic crystal has an elastic energy 2φ = Tc = c11 η2 and it is clear that c11 can be obtained as (∂2φ/∂η2). This particular deformation transforms the unit cell from a cube to a square-based prism of dimensions [a(1+η), a, a, 90, 90, 90], thus lowering the symmetry from cubic to tetragonal. The symmetry reduction can have the added consequence that some of the atoms within the cell gain degrees of freedom and their position within the cell should be reoptimized for each new value of η. Failing to take into account this inner strain can produce a significant impact on the calculated value of the elastic moduli. Example: Cubic CaF2 [fluorite; Fm¯ 3m (Num. 225); Ca (4a) (0, 0, 0); F (8c) (1/4, 1/4, 1/4)]. Strain Space G. Inner strain 2φ Cell η[1, 1, 0, 0, 0, 0] 4|mmm No 2(c11 +c12 )η2 [a(1+η), a(1+η), a, 90, 90, 90] η[1, 1, −2, 0, 0, 0] 4|mmm No 6(c11 −c12 )η2 [a(1+η), a(1+η), a(1−2η), 90, 90, 90] η[1, 1, 1, 0, 0, 0] Fm¯ 3m No 3(c11 +2c12 )η2 [a(1+η), a(1+η), a(1+η), 90, 90, 90] η[0, 0, 0, 1, 1, 1] R¯ 3m F (x, x, x) 3c44 η2 [a, a, a, α, α, α], 2η = cos(90−α) x ≈ 1/4 Ca and F remain on a symmetry fixed position for the first three proposed deformations, and F has only one degree of freedom in the fourth one. [η, η, η, 0, 0, 0] is a breathing dilatation of the cubic unit cell and, consequently, the elastic energy is proportional to the bulk modulus. The c44 modulus could also be obtained through a [0, 0, 0, 0, 0, η] deformation, but this would reduce the cell to moclinic symmetry, instead of rhombohedral as the proposed [0, 0, 0, η, η, η]. Designing a set of sensible deformations can be a creative task with a large influence on the computational effort. The best routes will elude creating inner strain and lowering the unit cell symmetry as much as possible. The bulk modulus, B, is related to the elastic moduli. In a cubic system, for instance, B = (c11 + 2c12 )/3. The importance of the elastic characterization. Bulk modulus and elastic moduli are very important properties of the crystal beyond their significant role on establishing the stability of the crystal phase and helping V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 78 / 86
textbooks Catti’s method II to predict the possible deformation of the unit cell shape in a potential phase transition. Elastic data is required in crystallography to calculate the thermal diffuse scattering correction to the Bragg diffraction intensities. The interpretation of the observed seismic waves, that constitute our best probe on Earth innards, depend on the assumed elastic properties of the solid phases present in the earth mantle. Material scientist are engaged in a long quest for superhard materials that maintain their hardness on a high pressure, high temperature, or high radiation working ambient. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 79 / 86
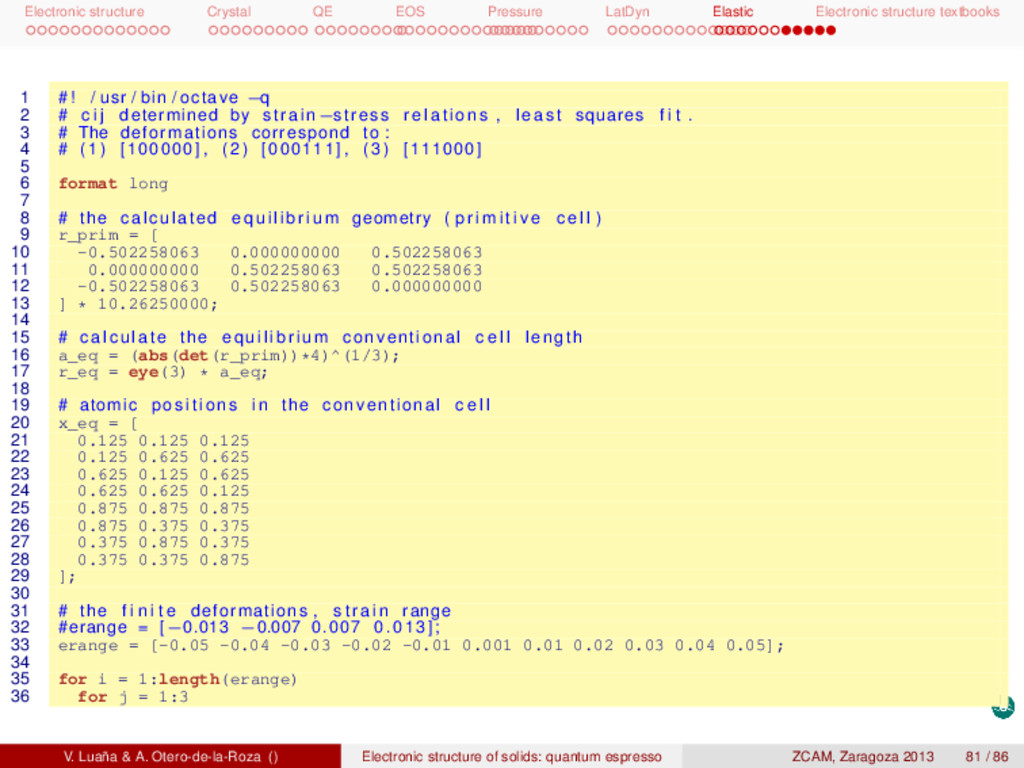
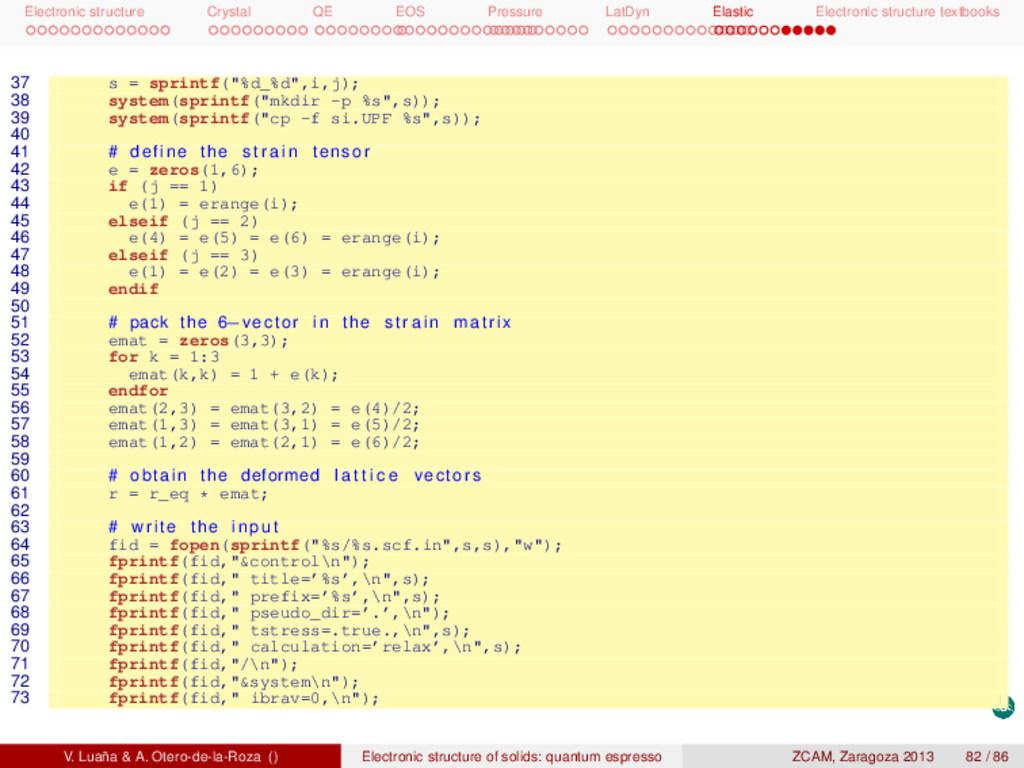
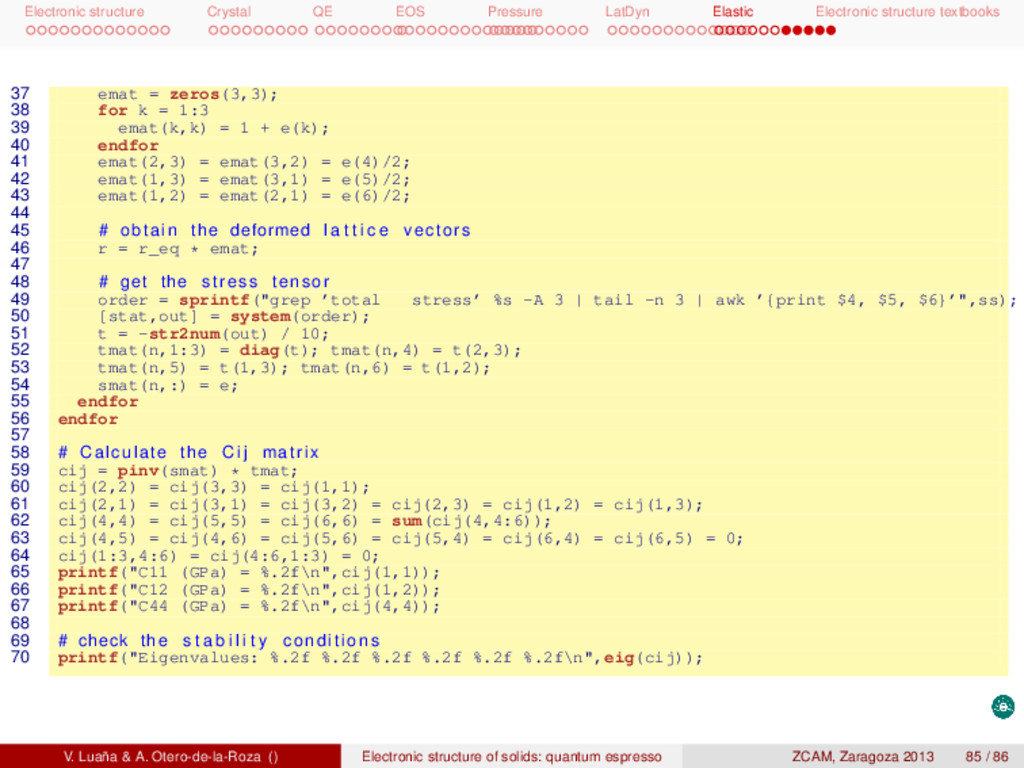
textbooks Elastic constants of Si Next scripts are designed to calculate the elastic constants of the diamond phase of Si. There are two main scripts: gen.m, that prepares the input to pwscf for all the calculations, and extract.m, that analyzes the pwscf outputs and produces the final results. Both scripts are written in octave, but extract.m calls the shell and combines the use of grep, tail, and awk. The whole calculation is done as 1 # ! / bin / bash −f 2 gen.m 3 for i in {1..11}_{1..3}/ ; do 4 cd $i 5 echo $i 6 pw.x < ${i%/}.scf.in > ${i%/}.scf.out 7 cd .. 8 done 9 extract.m The elastic constants are defined in terms of the conventional cubic cell, so we have performed all our calculations in this setting rather than using the rhombohedral primitive cell as in previous tasks. The calculation is based on three combined deformations: [111000], [001000], and [000111]. Several values of the deformations are performed on each case. Each point involves calculating the stress tensor (tstress=.true.) and relaxing the position of the ions (calculation=’relax’ and &ions/ namelist). The theoretical result reproduces well the experiments and describes a clearly stable phase. [GPa] c11 c12 c44 B range points calc. 154.4 56.8 75.9 89.3 ±0.013 4 calc. 153.9 57.4 75.6 89.6 ±0.050 11 expt. 165,6 63.9 79.5 97.8 We can observe that a particular least squares fitting is performed to determine the cij from the strees (τ) and strain ( ): pinv(strain)*stress, where prinv() is the Moore-Penrose pseudo inverse. As the argument to prinv() is not a square matrix, this function returns the least squares coefficients. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 80 / 86
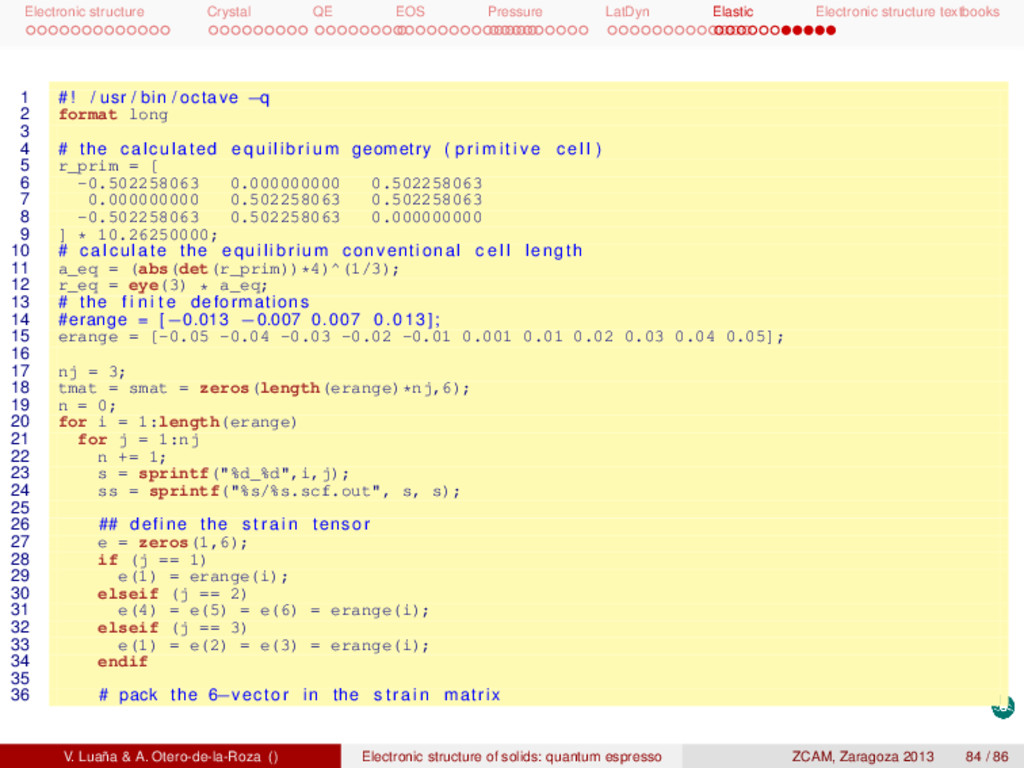
textbooks 1 # ! / usr / bin / octave −q 2 # c i j determined by strain−stress relations , least squares f i t . 3 # The deformations correspond to : 4 # (1) [100000] , (2) [000111] , (3) [111000] 5 6 format long 7 8 # the calculated equilibrium geometry ( p r i m i t i v e c e l l ) 9 r_prim = [ 10 -0.502258063 0.000000000 0.502258063 11 0.000000000 0.502258063 0.502258063 12 -0.502258063 0.502258063 0.000000000 13 ] * 10.26250000; 14 15 # calculate the equilibrium conventional c e l l length 16 a_eq = (abs(det(r_prim))*4)^(1/3); 17 r_eq = eye(3) * a_eq; 18 19 # atomic positions in the conventional c e l l 20 x_eq = [ 21 0.125 0.125 0.125 22 0.125 0.625 0.625 23 0.625 0.125 0.625 24 0.625 0.625 0.125 25 0.875 0.875 0.875 26 0.875 0.375 0.375 27 0.375 0.875 0.375 28 0.375 0.375 0.875 29 ]; 30 31 # the f i n i t e deformations , s t r a i n range 32 #erange = [−0.013 −0.007 0.007 0.013]; 33 erange = [-0.05 -0.04 -0.03 -0.02 -0.01 0.001 0.01 0.02 0.03 0.04 0.05]; 34 35 for i = 1:length(erange) 36 for j = 1:3 V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 81 / 86
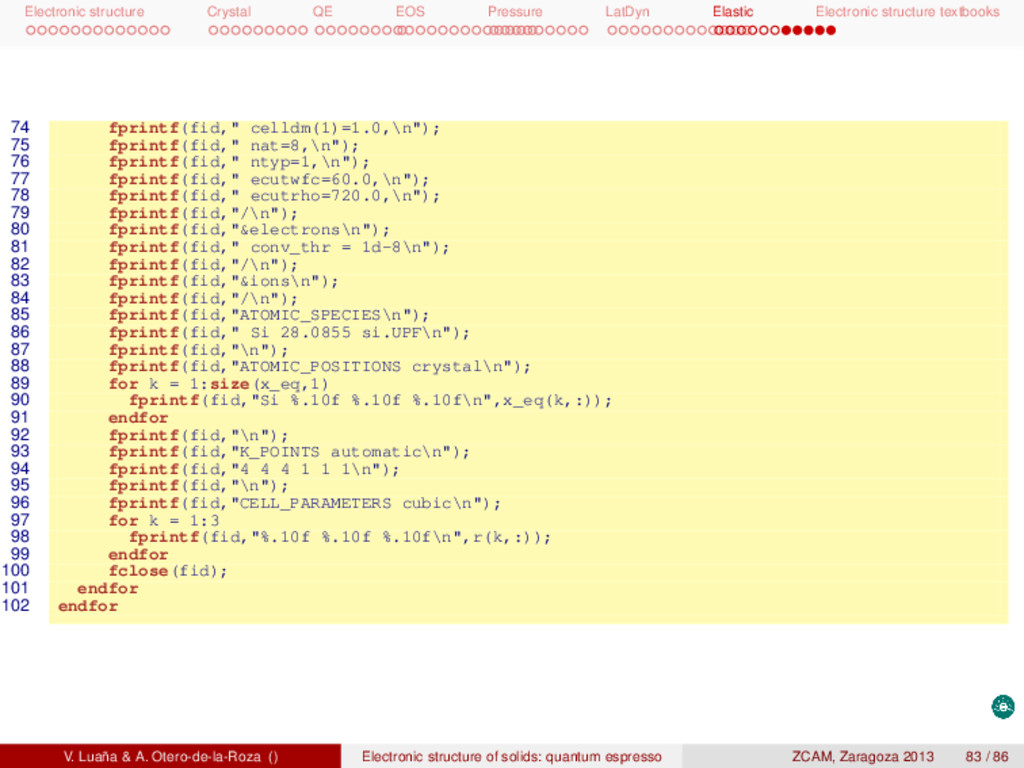
textbooks 1 # ! / usr / bin / octave −q 2 format long 3 4 # the calculated equilibrium geometry ( p r i m i t i v e c e l l ) 5 r_prim = [ 6 -0.502258063 0.000000000 0.502258063 7 0.000000000 0.502258063 0.502258063 8 -0.502258063 0.502258063 0.000000000 9 ] * 10.26250000; 10 # calculate the equilibrium conventional c e l l length 11 a_eq = (abs(det(r_prim))*4)^(1/3); 12 r_eq = eye(3) * a_eq; 13 # the f i n i t e deformations 14 #erange = [−0.013 −0.007 0.007 0.013]; 15 erange = [-0.05 -0.04 -0.03 -0.02 -0.01 0.001 0.01 0.02 0.03 0.04 0.05]; 16 17 nj = 3; 18 tmat = smat = zeros(length(erange)*nj,6); 19 n = 0; 20 for i = 1:length(erange) 21 for j = 1:nj 22 n += 1; 23 s = sprintf("%d_%d",i,j); 24 ss = sprintf("%s/%s.scf.out", s, s); 25 26 ## define the s t r a i n tensor 27 e = zeros(1,6); 28 if (j == 1) 29 e(1) = erange(i); 30 elseif (j == 2) 31 e(4) = e(5) = e(6) = erange(i); 32 elseif (j == 3) 33 e(1) = e(2) = e(3) = erange(i); 34 endif 35 36 # pack the 6−vector in the s t r a i n matrix V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 84 / 86
textbooks Electronic structure textbooks R. M. Martin, "Electronic Structure: Basic theory and practical methods" (Cam- bridge, 2004). L. N. Kantorovich, "Quantum Theory of the Solid State: An Introduction" (Kluwer, 2004). E. Kaxiras, "Atomic and Electronic Structure of Solids" (Cambridge, 2003). M. Marder, "Condensed Matter Physics" (Wiley-Interscience, 2000). [1] A. Otero-de-la Roza, V. Luaña, CRITIC2: topological analysis of scalar fields on crystals, Comput. Phys. Commun.(to be sub- mitted). [2] A. Otero-de-la Roza, V. Luaña, GIBBS2: A new version of the quasi-harmonic model code. I. Robust treatment of the static data, Comput. Phys. Commun. 182 (2011) 1708–1720. [3] M. A. Hopcroft, W. D. Nix, T. W. Kenny, What is the young’s modulus of silicon?, IEEE J. Microelectromechanical Systems 19 (2010) 229–238. [4] S. Bhagavantam, Crystal symmetry and physical properties, Academic, New York, 1966. [5] J. F. Nye, Physical Properties of Crystals, Oxford UP, Oxford, UK, 1985, republication of the 1957 classic. [6] M. Catti, Calculation of elastic constants by the method of crystal static deformation, Acta Cryst. A 41 (1985) 494–500. [7] M. Catti, Crystal elasticity and inner strain: A computational model, Acta Cryst. A 45 (1989) 20–25. V. Luaña & A. Otero-de-la-Roza () Electronic structure of solids: quantum espresso ZCAM, Zaragoza 2013 86 / 86
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}