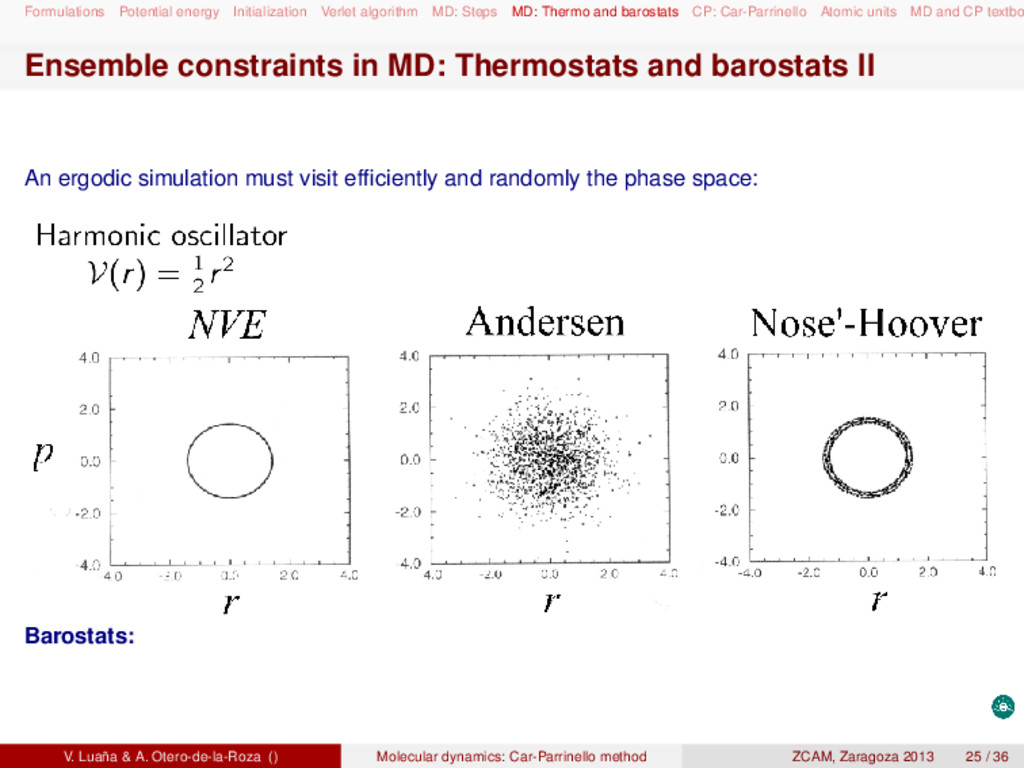
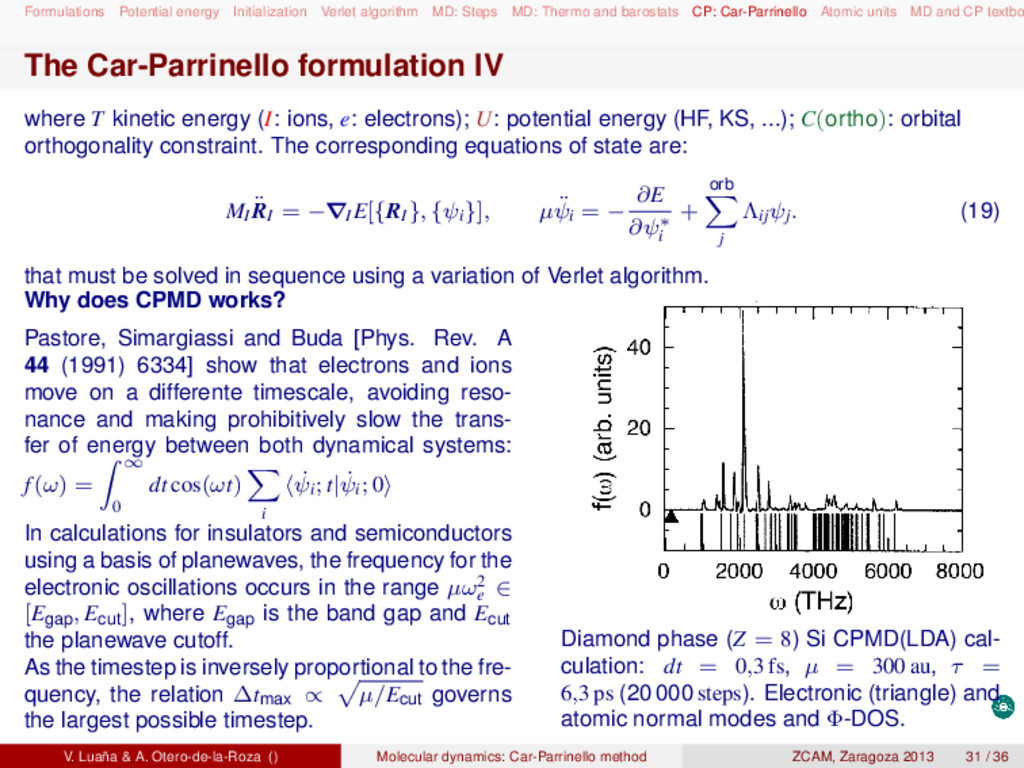
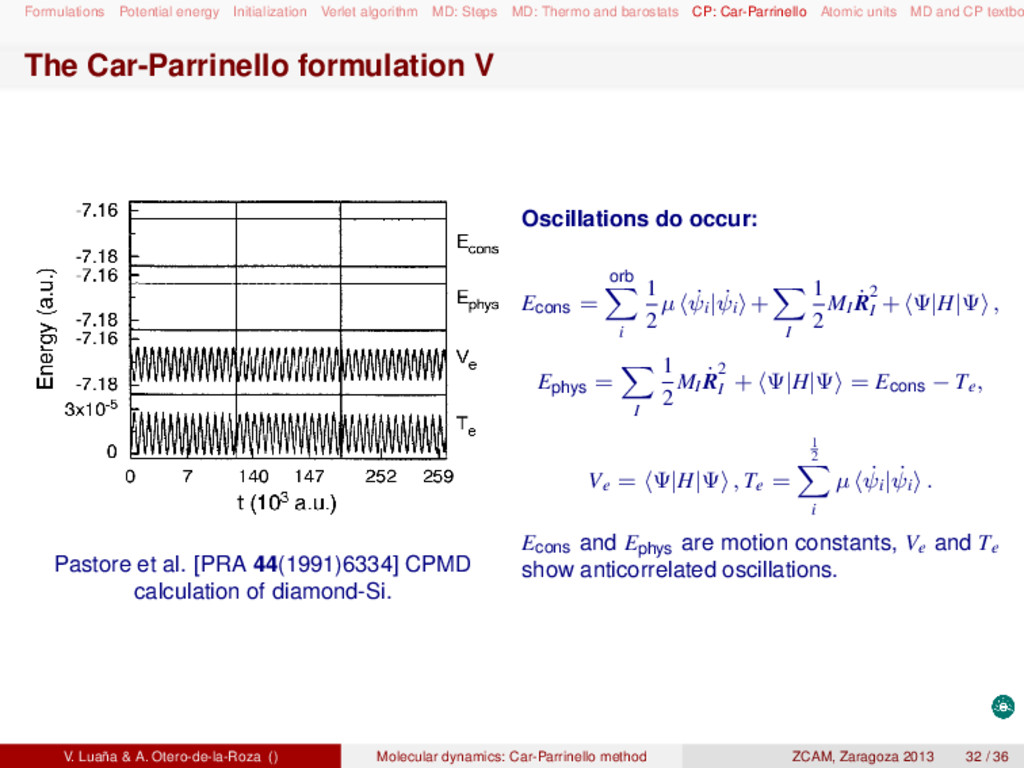
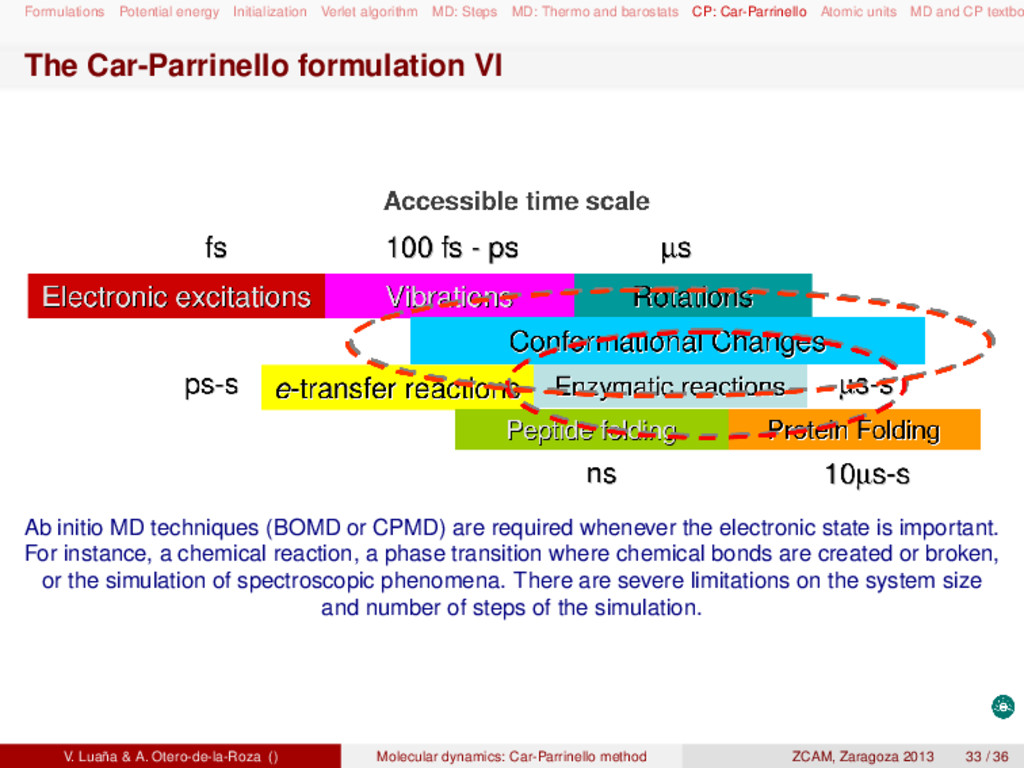
and barostats CP: Car-Parrinello Atomic units MD and CP textbo MD and CP textbooks Statistical thermodynamics: D. A. McQuarrie, Statistical mechanics (Harper Collins, 1976). D. Chandler, Introduction to modern statistical mechanics (Oxford UP, 1987). Molecular dynamics: M. P. Allen and D. J. Tildesley, Computer simulation of liquids (Clarendon Press, 1987). D. Frenkel and B. Smit, Understanding molecular simulation (Academic Press, 2005). Raugei Simone, Introduction to molecular dynamics simulations (people.sissa.it/~raugei/ lecture_notes/md_notes.pdf) G. Bussi, Theory and tips for molecular dynamics (sites.google.com/site/giovannibussi downloads/slides.pdf, 2008). Molecular simulations: A. R. Leach, Molecular modeling (Prentice Hall, 2001). T. Schlick, Molecular modeling and simulation (Springer, 2002). Car-Parrinello and ab initio molecular dynamics: D. Marx and J. Hutter, "Ab initio molecular dynamics: Theory and implementation", in Mod- ern methods and algorithms on quantum chemistry, J. Grotendorst (Ed.), (John von Neu- mann Institute, NIC series vol. 1 & 3, 2000) http://www2.fz-juelich.de/nic-series/ Volume3/marx.pdf. Raugei Simone, Introduction to Car-Parrinello molecular dynamics simulations (http://people sissa.it/~raugei/lecture_notes/cp.pdf) V. Luaña & A. Otero-de-la-Roza () Molecular dynamics: Car-Parrinello method ZCAM, Zaragoza 2013 36 / 36
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}