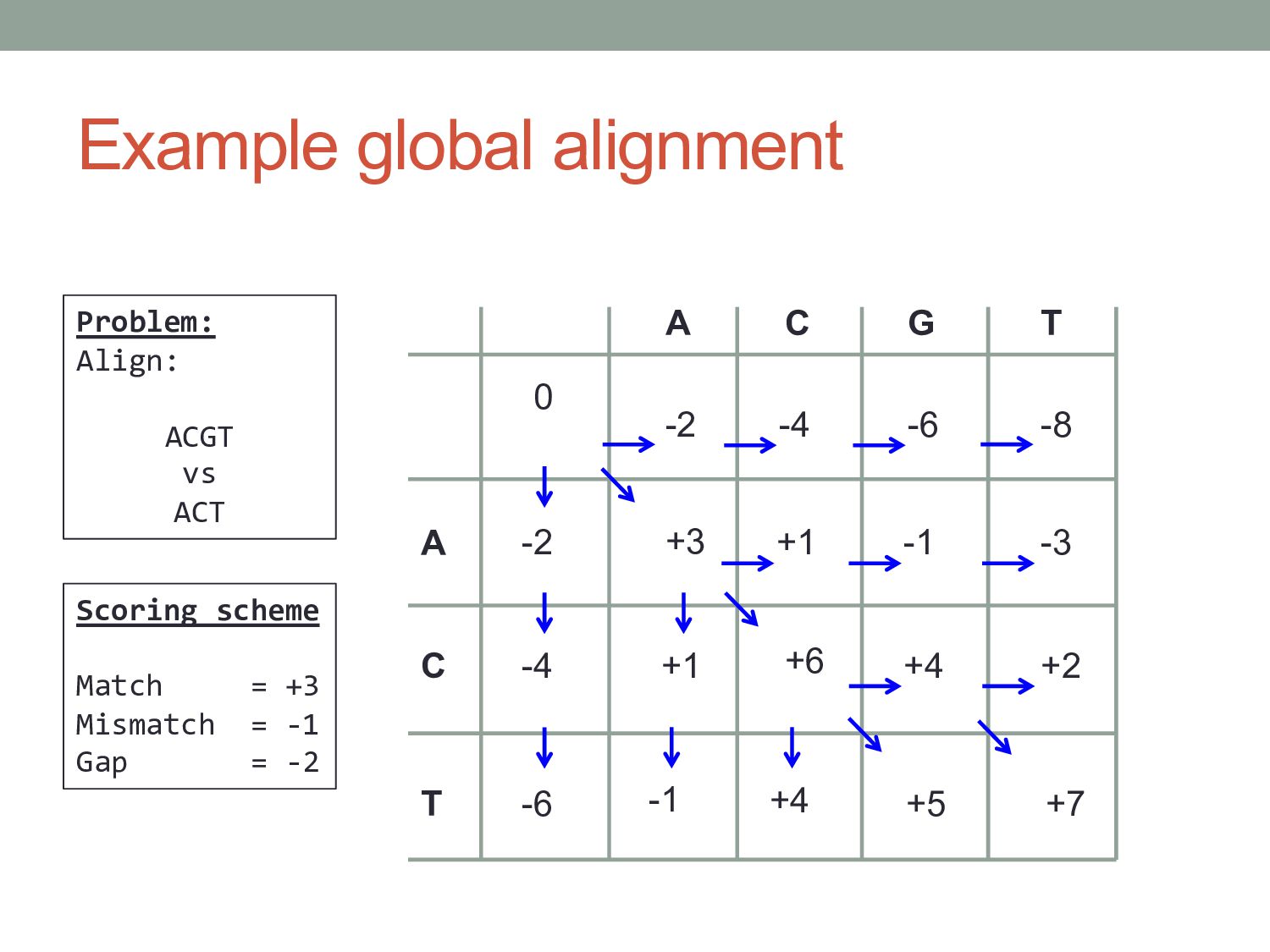
j Fi, j-1 Fi, j -d -d s(X i , Y j ) Let: Fi, j = optimal score of aligning X1 …Xi to Y1 …Yj Three possibilities: • Xi aligns to Yj : Fi, j = Fi-1, j-1 + s(Xi , Yj ) • Xi aligns to gap : Fi, j = Fi-1, j – d • Yj aligns to gap : Fi, j = Fi, j-1 – d
j * d F1…i, 0 = - i * d for each i = 1…M for each j = 1…N Fi-1, j-1 + s(Xi , Yj ) [match] Fi, j = max Fi-1, j – d [gap in X] Fi, j-1 – d [gap in Y] DIAG, if [match] Ptri, j = LEFT, if [gap in X] UP, if [gap in Y] Initialization Iteration Termination: FM, N is the score of the optimal alignment. Alignment path can be traced back from PtrM, N Problem: Find the best way to align X and Y from 0,0 to M,N
Brute force alignment: • Possible pairwise alignments: • Needleman-Wunsch alignment: • 3 summations and a max operation per matrix entry • L x L matrix entries to compute • à O(L2) 22L 2πL
X and Y. Given X1 …XM and Y1 …YN , find i, j, k, l such that the score of alignment between Xi …Xj and Yk …Yl is maximal. Idea: If the alignment score becomes negative, it is better to start a new alignment. i.e. set the score to 0
F1…i, 0 = 0 for each i = 1…M for each j = 1…N Fi-1, j-1 + s(Xi , Yj ) [match] Fi, j = max Fi-1, j – d [gap in X] Fi, j-1 – d [gap in Y] 0 DIAG, if [match] Ptri, j = LEFT, if [gap in X] UP, if [gap in Y] Initialization Iteration Termination: Best local alignment score is the Fi, j with maximum value. Best local alignment path can be traced back from Ptri, j corresponding to maximum Fi, j
Seq1 AAGACCTACGGGGGGGGTTCCCGCACTTCGACCTGAGCC Seq2 AAGACCTACG-G-G-G-TTCCCGCACTTCGACCTGAGCC Alignment 1 Seq1 AAGACCTACGGGGGGGGTTCCCGCACTTCGACCTGAGCC Seq2 AAGACCTACGGGG----TTCCCGCACTTCGACCTGAGCC Alignment 2 How many evolutionary events does each alignment suggest? Should gap opening ”cost” the same as gap extension?
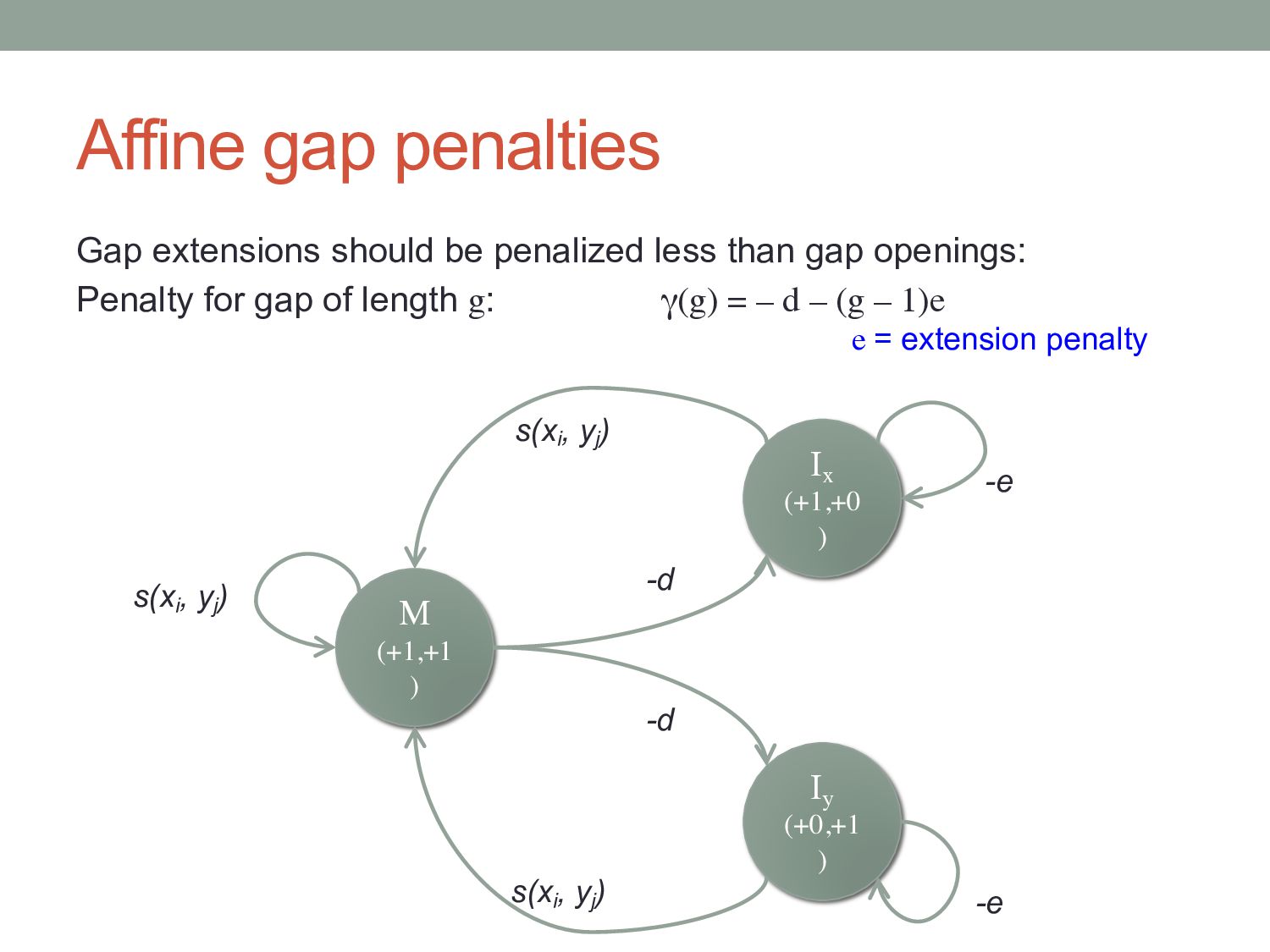
gap openings: Penalty for gap of length g: γ(g) = – d – (g – 1)e To implement, we need two more matrices to keep track of insertions: Fi, j = max{ Mi, j , Ixi, j , Iyi, j } Mi-1, j-1 + s(Xi , Yj ) Mi, j = max Ixi-1, j-1 + s(Xi , Yj ) Iyi-1, j-1 + s(Xi , Yj ) Mi-1, j – d Ixi, j = max Ixi-1, j – e Mi, j-1 – d Iyi, j = max Iyi, j-1 – e e = extension penalty
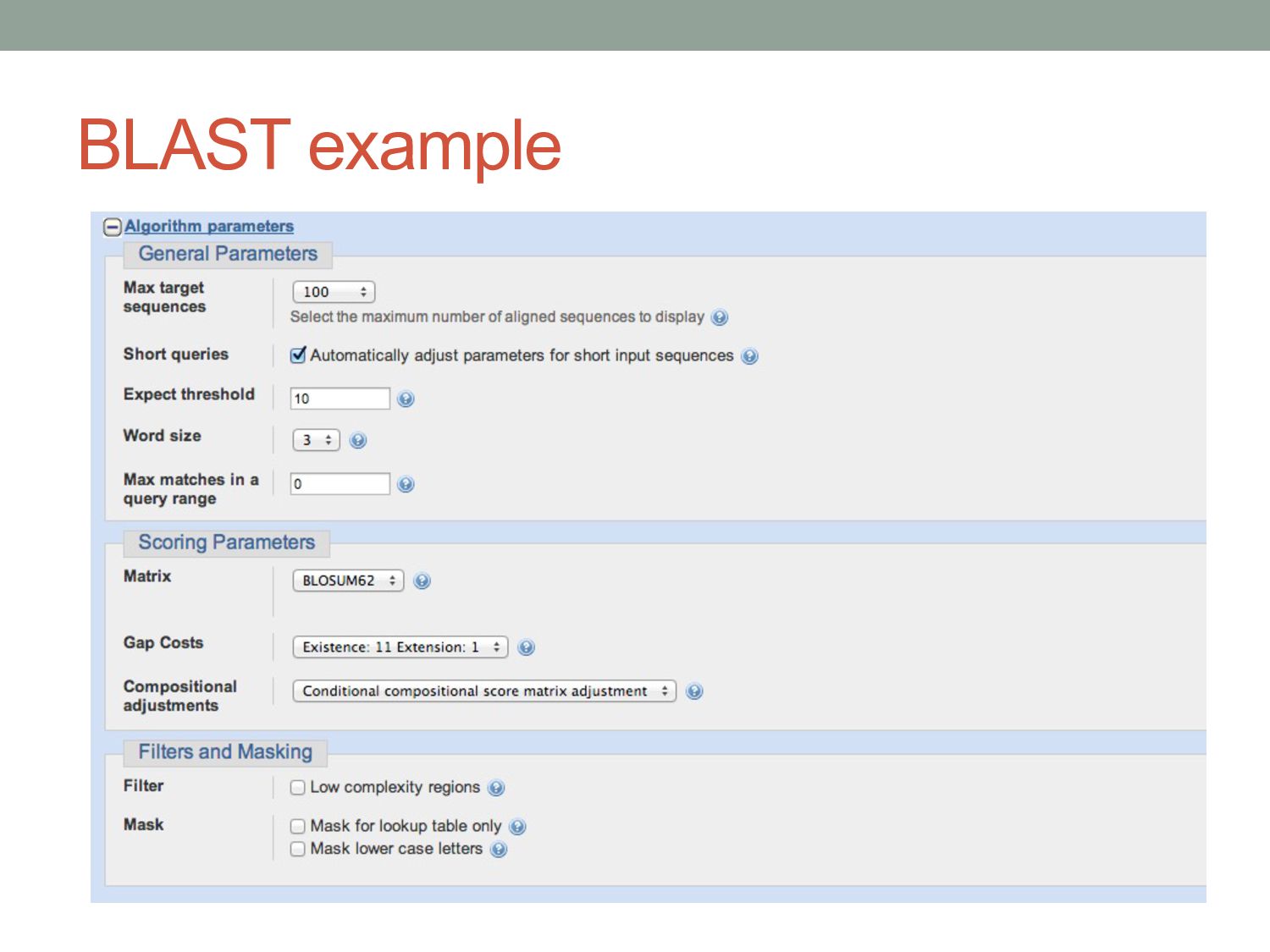
Yj ) A C G T A +1 -2 -2 -2 C -2 +1 -2 -2 G -2 -2 +1 -2 T -2 -2 -2 +1 Default nucleotide substitution matrix used by BLAST A ratio of 0.33 (1/-3) is appropriate for sequences that are about 99% conserved; a ratio of 0.5 (1/-2) is best for sequences that are 95% conserved; a ratio of about one (1/-1) is best for sequences that are 75% conserved (https://www.ncbi.nlm.nih.gov/books/NBK279678) Gap open penalty = -5 Gap extension penalty = -2
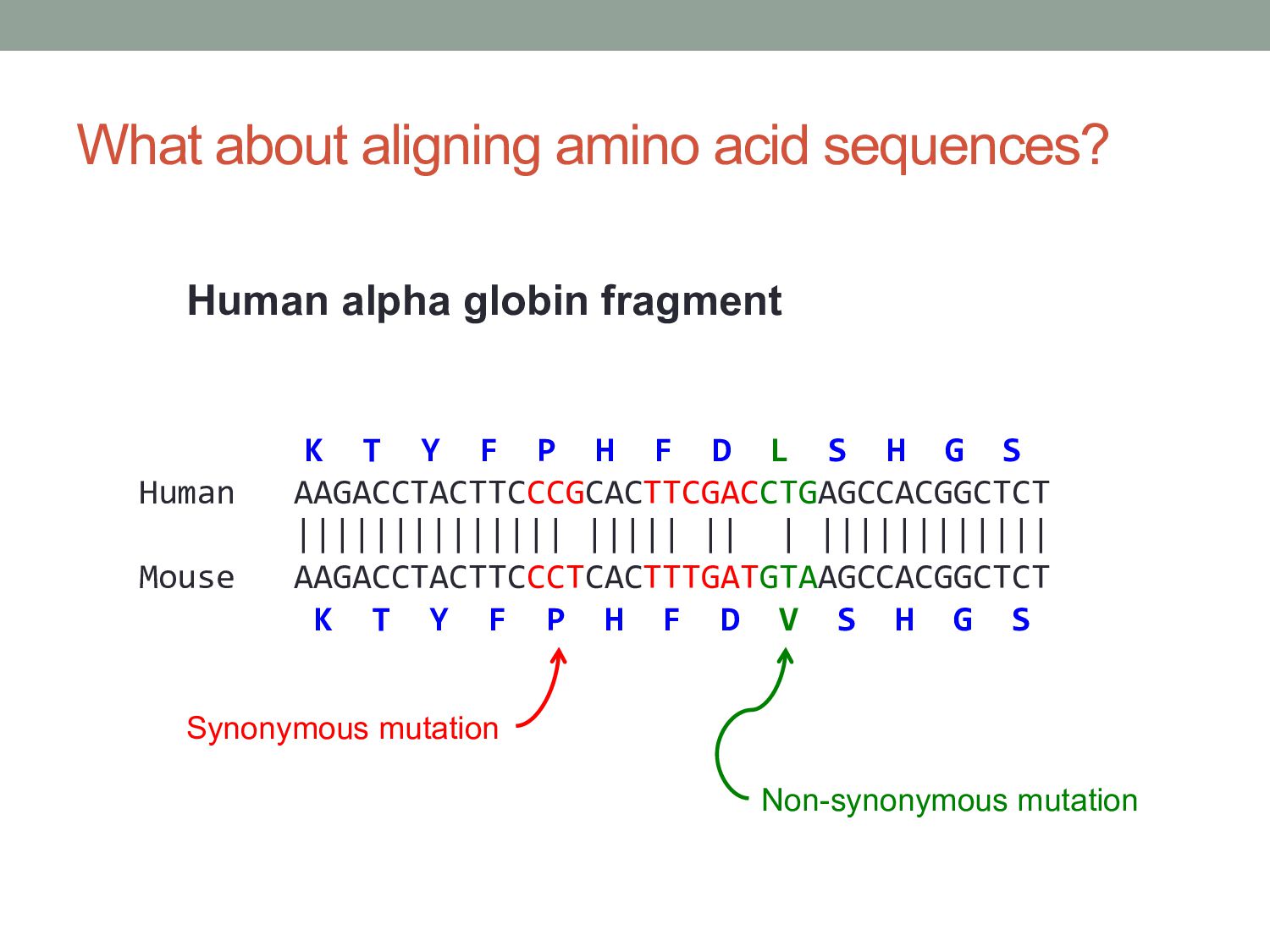
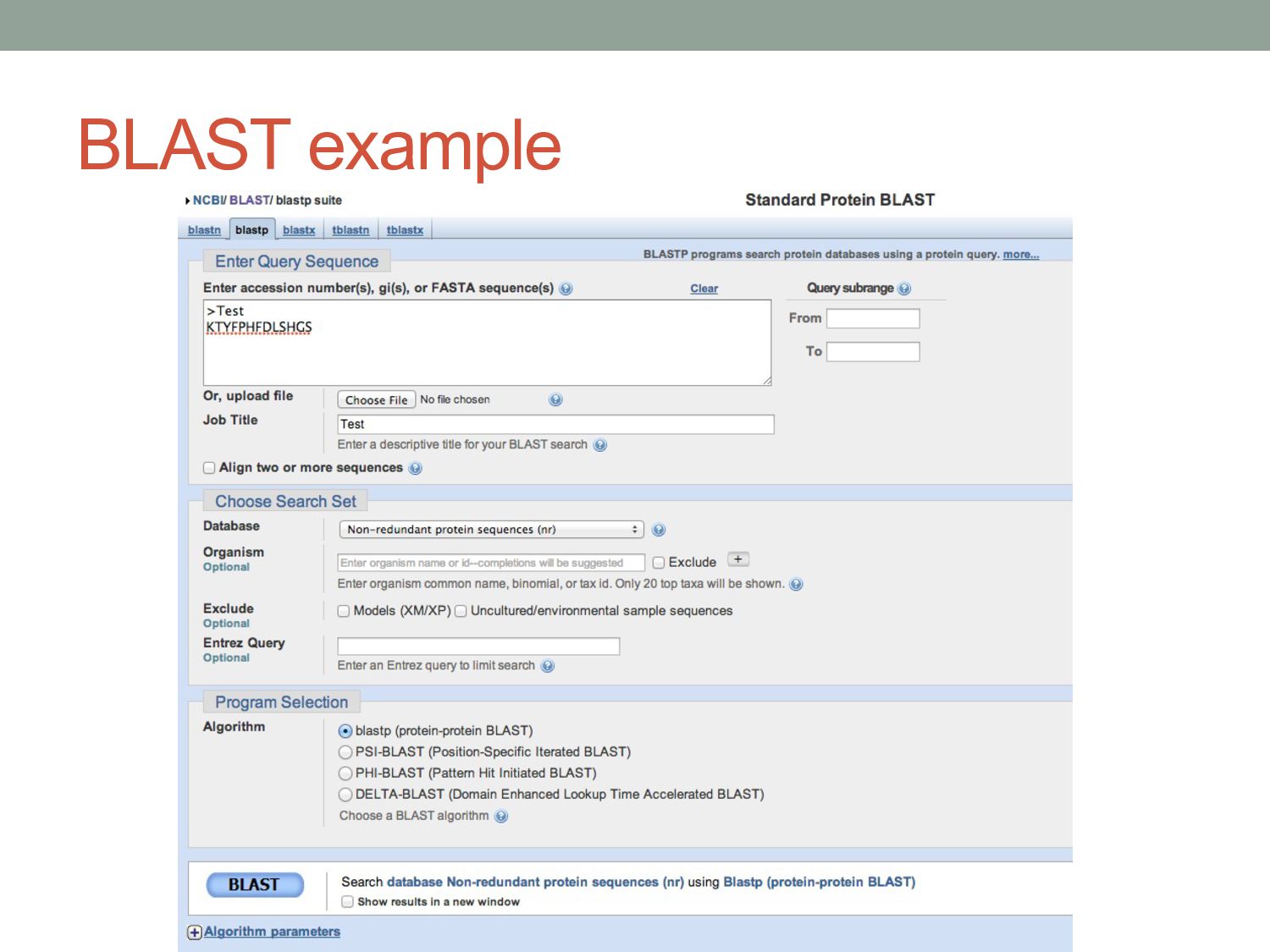
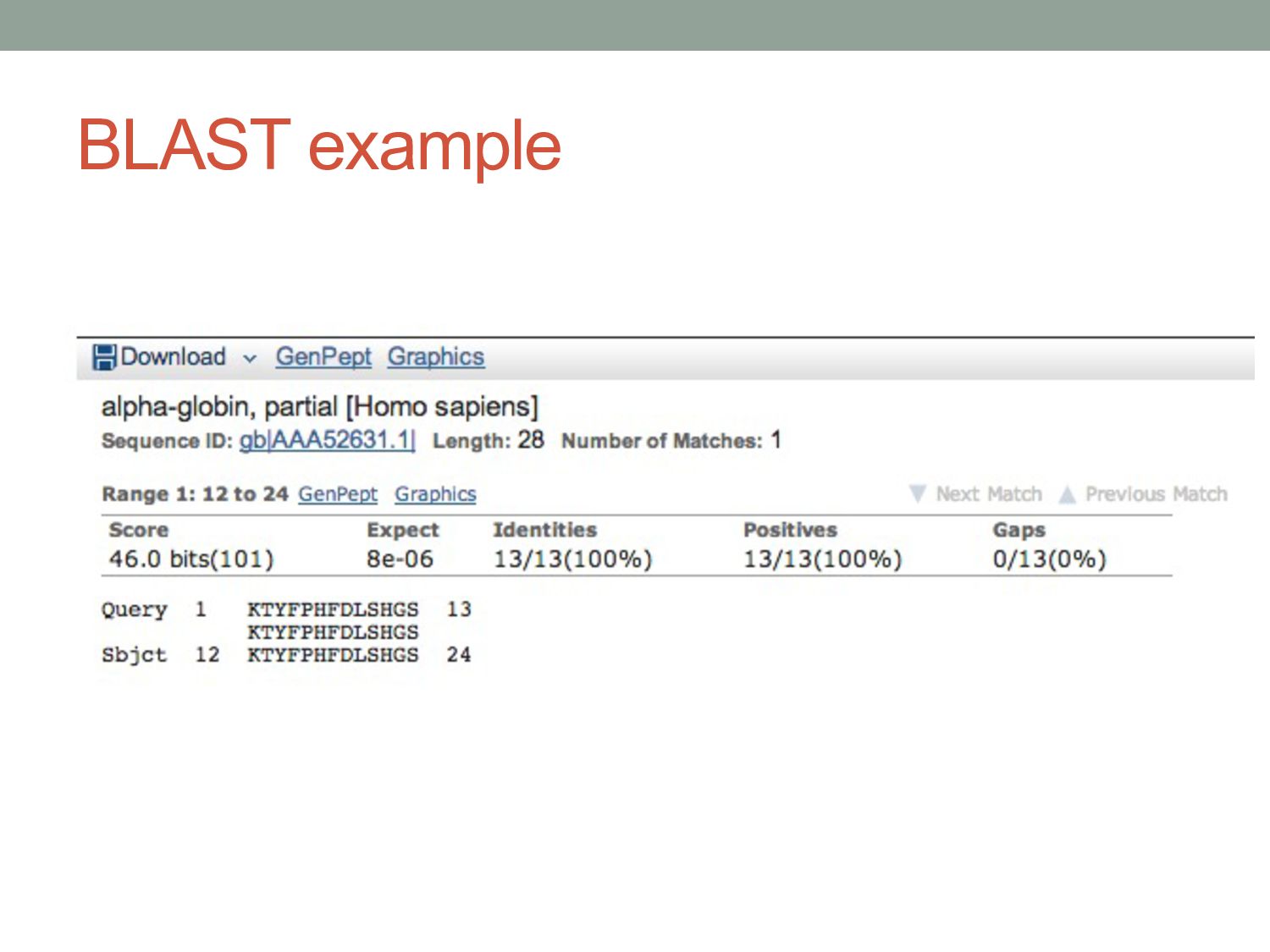
P H F D L S H G S Human AAGACCTACTTCCCGCACTTCGACCTGAGCCACGGCTCT |||||||||||||| ||||| || | |||||||||||| Mouse AAGACCTACTTCCCTCACTTTGATGTAAGCCACGGCTCT K T Y F P H F D V S H G S Human alpha globin fragment Synonymous mutation Non-synonymous mutation
probability that two residues would appear at equivalent/aligned positions in homologous sequences. • Likelihood ratio between two hypotheses: • Hypothesis 1: residues are aligned due to chance • unrelated sequences • Hypothesis 2: residues are aligned due to common ancestry • evolutionarily related sequences
similar sequences are homologous • i.e. related via evolution • Likelihood ratio between two hypotheses: • Hypothesis 1: alignment due to chance (unrelated sequences) • Hypothesis 2: alignment due to common ancestry (related sequences) • Calculate probability of observing an alignment according to each hypothesis • P(Xi ,Yj | U): prob of aligning Xi & Yj by model U (unrelated) • P(Xi ,Yj | R): prob of aligning Xi & Yj by model R (related) • Alignment score: likelihood ratio between the two • Relative likelihood that alignment not due to chance = P(Xi ,Yj | R) / P(Xi ,Yj | U) • Score = log(P)
the alignment is randomly generated according to some amino acid frequencies. • Let qa be the probability of seeing amino acid a. • Probability of seeing an n-character alignment of X & Y: P(X ,Y |U) = q Xi i=1 n ∏ q Yi i=1 n ∏
amino acids evolved from a common ancestor. • Let pa,b be the probability that evolution gave rise to amino acid a in one sequence and amino acid b in the other sequence. • Define from known high-quality alignments of related protein sequences. • Probability of seeing an n-character alignment of X & Y: P(X,Y | R) = p Xi ,Yi i=1 n ∏
explains the alignment (U or R)? • Consider the relative likelihood of two possibilities (log-odds): P(X,Y | R) P(X,Y |U) = p Xi ,Yi i=1 n ∏ q Xi i=1 n ∏ q Yi i=1 n ∏ log P(X,Y | R) P(X,Y |U) = log p Xi ,Yi q Xi q Yi i n ∑
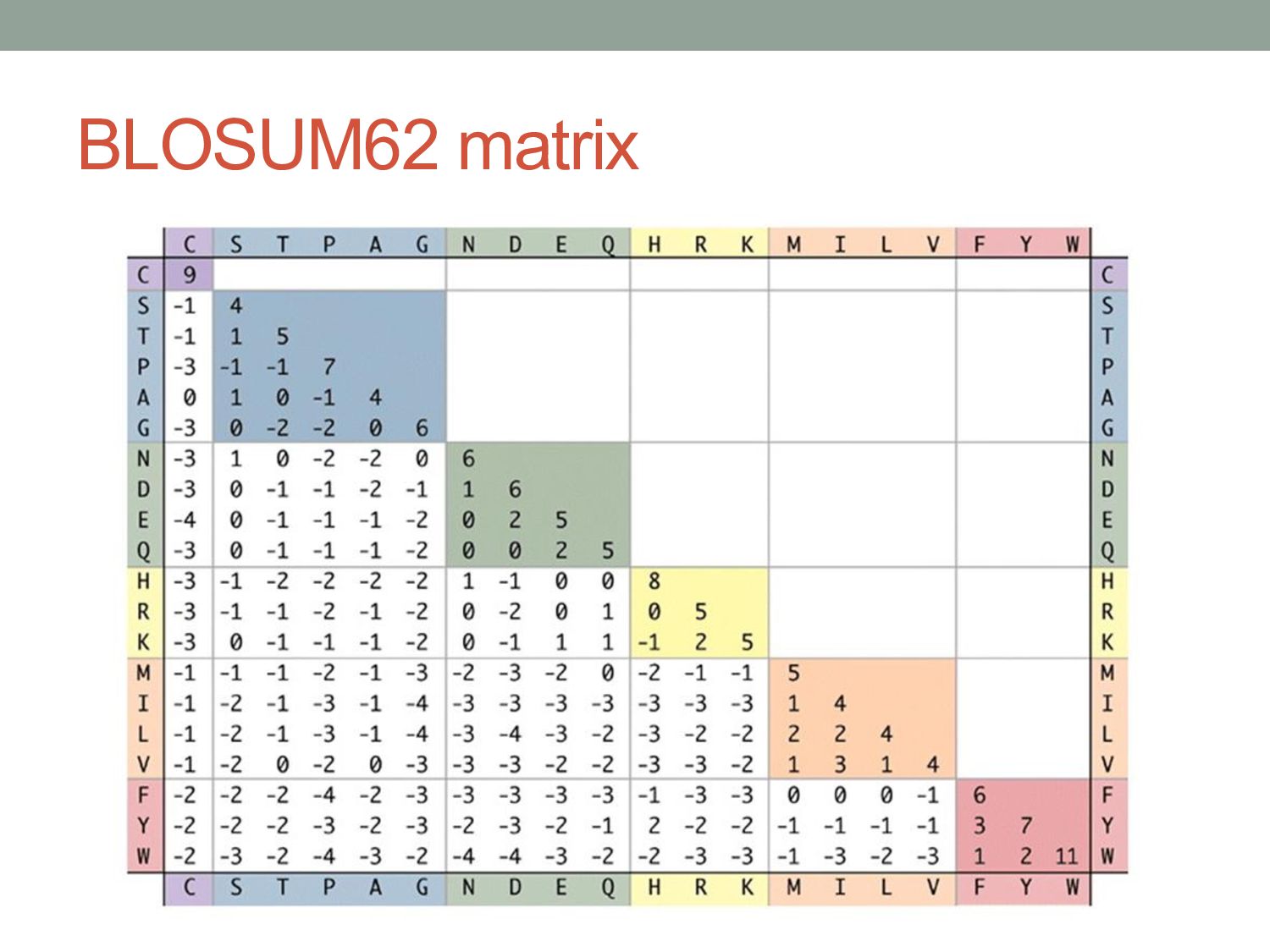
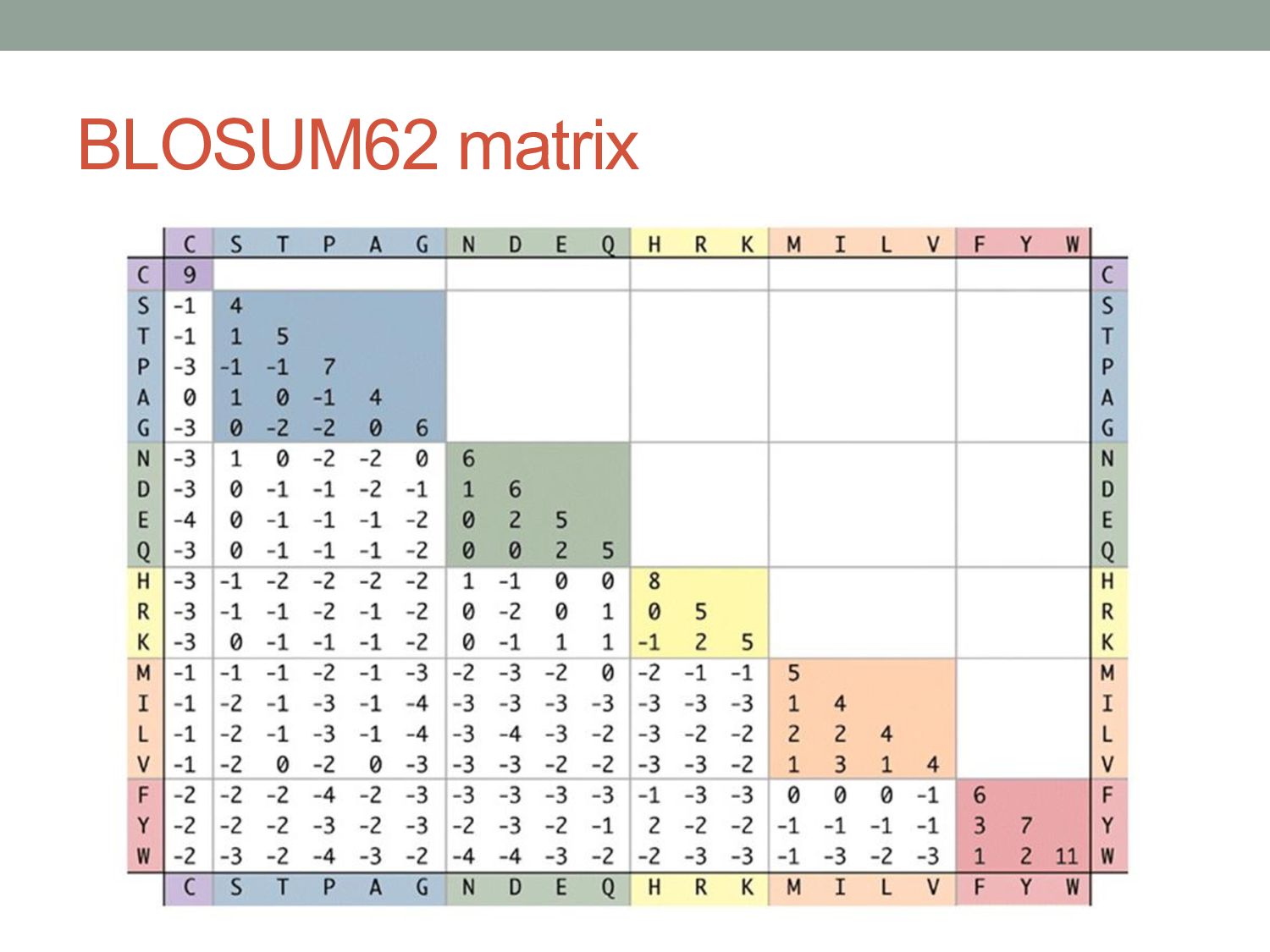
alignments of many homologous proteins. • PAM matrices (“point accepted mutations”): • Began with closely related protein sequences (>85% identical). • Built phylogenetic trees to model evolutionary distances. • BLOSUM (“blocks substitution matrix”): • Based on ungapped alignments from BLOCKS database of related proteins. • Calculated substitution frequencies between related proteins with more than X% identity (e.g. X = 80% for BLOSUM80). BLOSUM80 PAM1 BLOSUM62 PAM120 BLOSUM45 PAM250 Less divergent More divergent
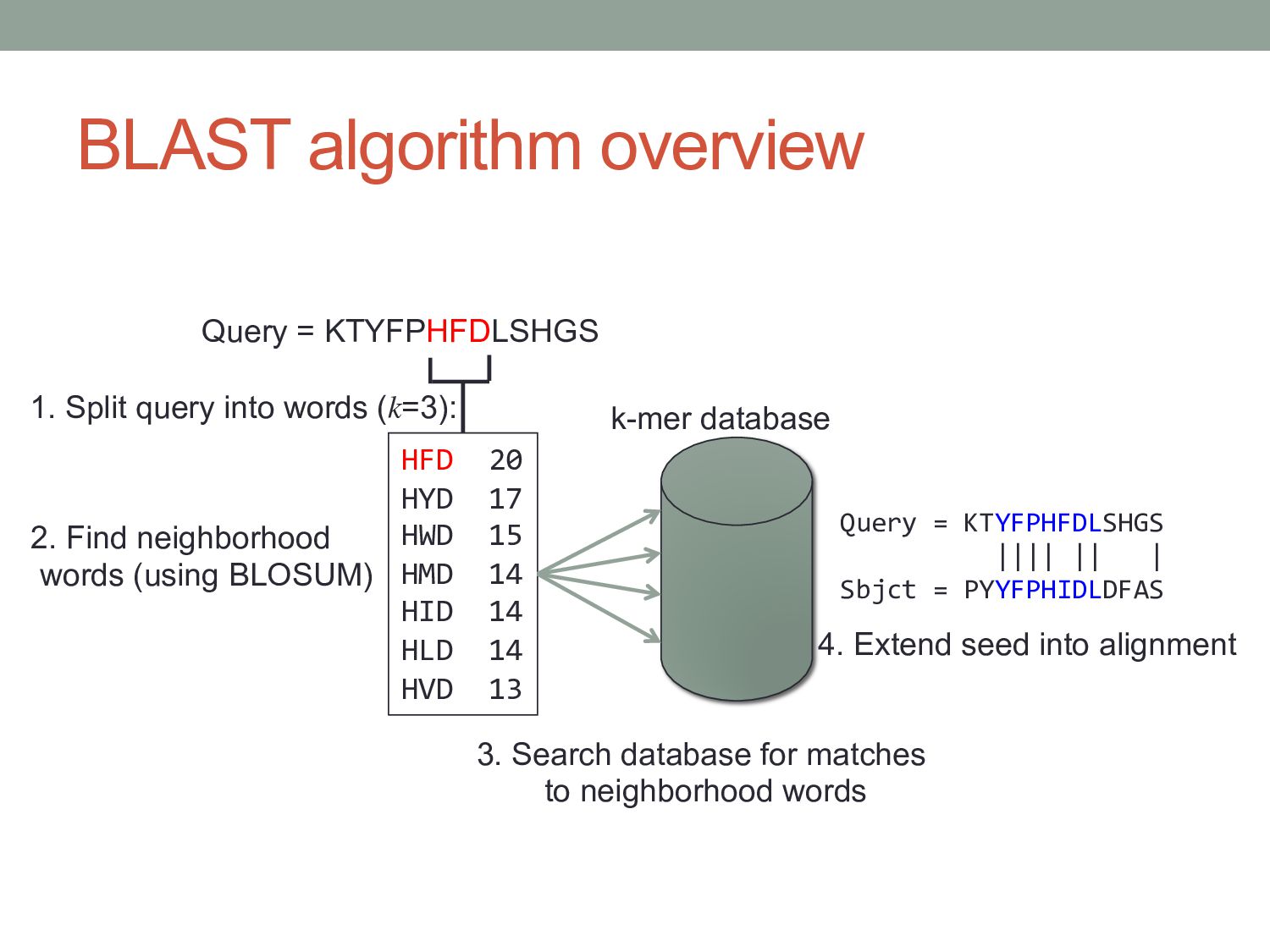
likely to contain a short stretch of very high scoring matches 1. Split query into overlapping words of length k. k = 20 for DNA, k = 6 for amino acids 2. Find neighborhood words for each k-mer. 3. Look up sequences that contain neighborhood words from table (seeds S). 4. Extend seeds S until score drops off. 5. Report significance and alignment of each match.
alignment problem, each applicable to different biological questions/assumptions. • Substitution matrices provide sequence similarity scores that account for some of the ways that sequences evolve.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}