1117-1123 originally published online February 27, 2009 Genome Res. Jared T. Simpson, Kim Wong, Shaun D. Jackman, et al. ABySS: A parallel assembler for short read sequence data Material Supplemental http://genome.cshlp.org/content/suppl/2009/04/27/gr.089532.108.DC1.html References http://genome.cshlp.org/content/19/6/1117.full.html#related-urls Article cited in: http://genome.cshlp.org/content/19/6/1117.full.html#ref-list-1 This article cites 31 articles, 14 of which can be accessed free at: Open Access Freely available online through the Genome Research Open Access option. Related Content Genome Res. December 7, 2011 : Jared T Simpson and Richard Durbin structures Efficient de novo assembly of large genomes using compressed data Genome Res. December 6, 2011 : Steven L Salzberg, Adam M Phillippy, Aleksey V Zimin, et al. GAGE: A critical evaluation of genome assemblies and assembly algorithms service Email alerting click here top right corner of the article or Receive free email alerts when new articles cite this article - sign up in the box at th Cold Spring Harbor Laboratory Press on January 4, 2012 - Published by genome.cshlp.org Downloaded from ARTICLE OPEN doi:10.1038/nature12211 The Norway spruce genome sequence and conifer genome evolution Lists of authors and their affiliations appear at the end of the paper Conifers have dominated forests for more than 200 million years and are of huge ecological and economic importance. Here we present the draft assembly of the 20-gigabase genome of Norway spruce (Picea abies), the first available for any gymnosperm. The number of well-supported genes (28,354) is similar to the .100 times smaller genome of Arabidopsis thaliana, and there is no evidence of a recent whole-genome duplication in the gymnosperm lineage. Instead, the large genome size seems to result from the slow and steady accumulation of a diverse set of long-terminal repeat transposable elements, possibly owing to the lack of an efficient elimination mechanism. Comparative sequencing of Pinus sylvestris, Abies sibirica, Juniperus communis, Taxus baccata and Gnetum gnemon reveals that the transposable element diversity is shared among extant conifers. Expression of 24-nucleotide small RNAs, previously implicated in transposable element silencing, is tissue-specific and much lower than in other plants. We further identify numerous long (.10,000 base pairs) introns, gene-like fragments, uncharacterized long non-coding RNAs and short RNAs. This opens up new genomic avenues for conifer forestry and breeding. Gymnosperms are a group of land plants comprising the extant taxa, cycads,Ginkgo, gnetophytes and conifers. Gymnospermsfirst appeared more than300 million years ago (Myrago)1, wellbefore theangiosperm lineage separated from the stem group of extant gymnosperms2. The negates the production of inbred lines that could facilitate genome assembly. The availability of conifer genome sequences would enable com- parative analyses of genome architecture and the evolution of key Vol. 29 no. 12 2013, pages 1492–1497 BIOINFORMATICS ORIGINAL PAPER doi:10.1093/bioinformatics/btt178 Genome analysis Advance Access publication May 22, 2013 Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data Inanc Birol1,2,3,*, Anthony Raymond1, Shaun D. Jackman1, Stephen Pleasance1, Robin Coope1, Greg A. Taylor1, Macaire Man Saint Yuen4, Christopher I. Keeling4, Dana Brand1, Benjamin P. Vandervalk1, Heather Kirk1, Pawan Pandoh1, Richard A. Moore1, Yongjun Zhao1, Andrew J. Mungall1, Barry Jaquish5, Alvin Yanchuk5, Carol Ritland4,6, Brian Boyle7, Jean Bousquet7,8, Kermit Ritland6, John MacKay7,8, Jo ¨ rg Bohlmann4,6 and Steven J.M. Jones1,2,9 1Genome Sciences Centre, British Columbia Cancer Agency, Vancouver, BC V5Z 4S6, Canada, 2Department of Medical Genetics, University of British Columbia, Vancouver, BC V6H 3N1, Canada, 3School of Computing Science, Simon Fraser University, Burnaby, BC V5A 1S6, Canada, 4Michael Smith Laboratories, University of British Columbia, Vancouver, BC V6T 1Z4, Canada, 5British Columbia Ministry of Forests, Lands and Natural Resource Operations, Victoria, BC V8W 9C2, Canada, 6Department of Forest Sciences, University of British Columbia, Vancouver, BC V6T 1Z4, Canada, 7Institute for Systems and Integrative Biology, Universite ´ Laval, Que ´ bec, QC G1K 7P4, Canada, 8Department of Wood and Forest Sciences, Universite ´ Laval, Que ´ bec, QC G1V 0A6, Canada and 9Department of Molecular Biology and Biochemistry, Simon Fraser University, Burnaby, BC V5A 1S6, Canada Associate Editor: Michael Brudno ABSTRACT White spruce (Picea glauca) is a dominant conifer of the boreal forests of North America, and providing genomics resources for this commer- cially valuable tree will help improve forest management and conser- vation efforts. Sequencing and assembling the large and highly repetitive spruce genome though pushes the boundaries of the current technology. Here, we describe a whole-genome shotgun sequencing strategy using two Illumina sequencing platforms and an assembly approach using the ABySS software. We report a 20.8 giga base pairs draft genome in 4.9 million scaffolds, with a scaffold N50 of 20356bp. We demonstrate how recent improvements in the sequen- cing technology, especially increasing read lengths and paired end reads from longer fragments have a major impact on the assembly contiguity. We also note that scalable bioinformatics tools are instru- mental in providing rapid draft assemblies. Availability: The Picea glauca genome sequencing and assembly data are available through NCBI (Accession#: ALWZ0100000000 PID: PRJNA83435). http://www.ncbi.nlm.nih.gov/bioproject/83435. Contact:
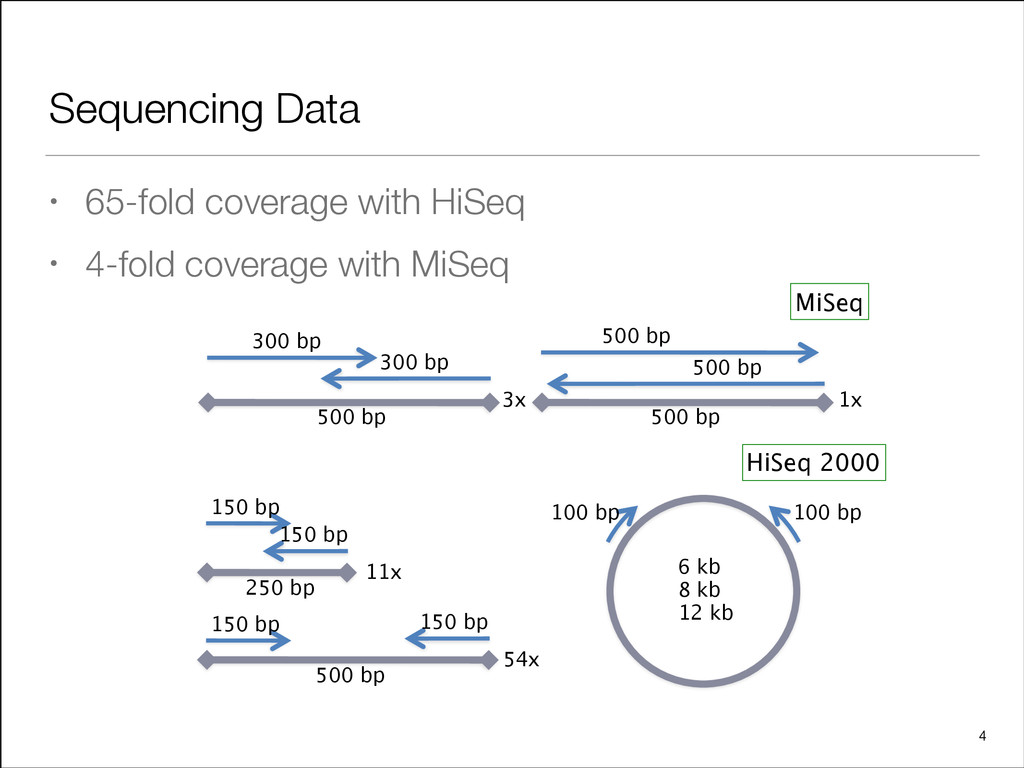
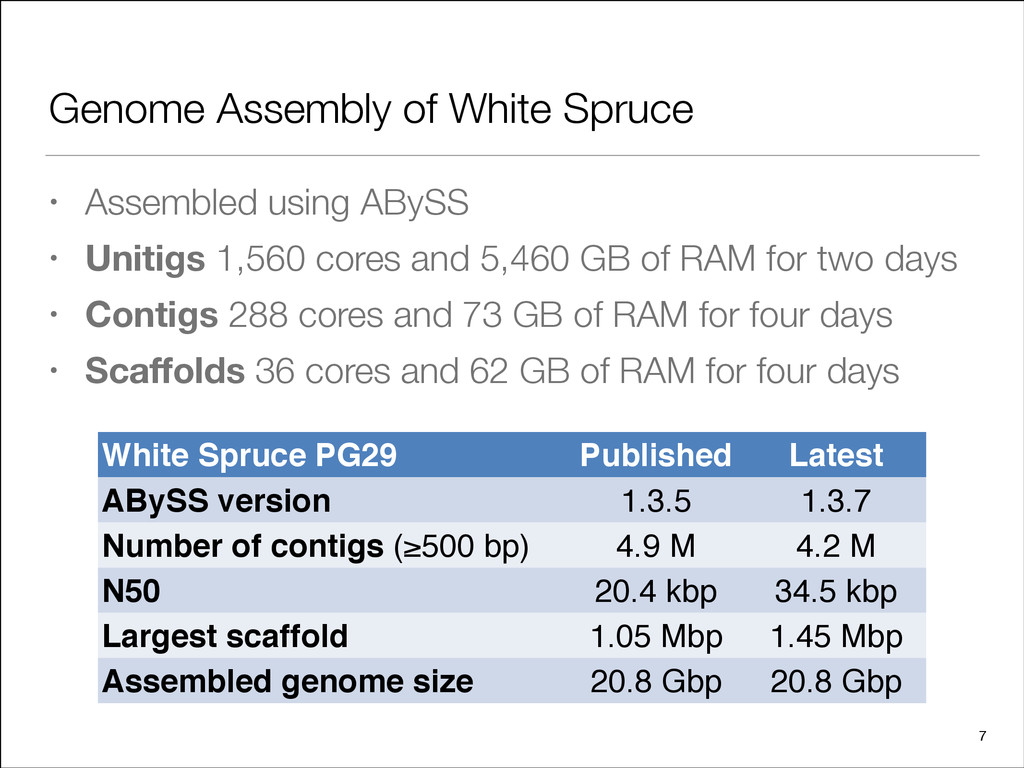
[email protected] Supplementary information: Supplementary data are available at Bioinformatics online. Received on March 20, 2013; revised on April 10, 2013; accepted on April 11, 2013 1 INTRODUCTION The assembly of short reads to develop genomic resources for non-model species remains an active area of development (Schatz et al., 2012). The feasibility of the approach and its scalability to large genomes was demonstrated by the ABySS publication (Simpson et al., 2009) using human genome sequencing data and was later used to assemble the panda genome with the SOAPdenovo tool (Li et al., 2010). The technology provides high quality results, as demonstrated for bacteria (Bankevich et al., 2012; Ladner et al., 2013; Ribeiro et al., 2012), and has been successfully applied numerous times on more complex gen- omes (Chan et al., 2011; Chu et al., 2011; Diguistini et al., 2009, 2011; Godel et al., 2012; Swart et al., 2012). Estimated at 20 giga base pairs (Gb) (Murray, 1998), sequen- cing and assembly of the genome of this gymnosperm species of the pine (Pinaceae) family present unique challenges. On the data generation end, those challenges include representation biases in whole-genome shotgun sequencing data, and difficulties in build- ing reduced representation resources to scale down the magni- tude of the problem. On the bioinformatics end, assembling massive sequencing datasets is extremely demanding on comput- ing cycles, memory usage, storage requirements, and for parallel programming implementations on communication traffic. We addressed the data representation challenges by preparing and sequencing multiple whole-genome shotgun libraries on the HiSeq 2000 and MiSeq sequencers from Illumina (San Diego, CA, USA). Compared with localized sequencing protocols, such as building and sequencing fosmid libraries, or the recent approach of isolating $10 kb DNA strands to generate indexed sequencing fragments in high throughput (Moleculo, San Diego, CA, USA), a shotgun only sequencing approach rapidly provides sequence data effectively covering the target genome at a cost that can be an order of magnitude less. The difference in cost is especially substantial when sequencing a large genome. In this work, we demonstrate that shotgun sequence assembly at this scale remains viable and produces valuable results. To *To whom correspondence should be addressed. ß The Author 2013. Published by Oxford University Press. at University of British Columbia on September 6, 2013 http://bioinformatics.oxfordjournals.org/ Downloaded from
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
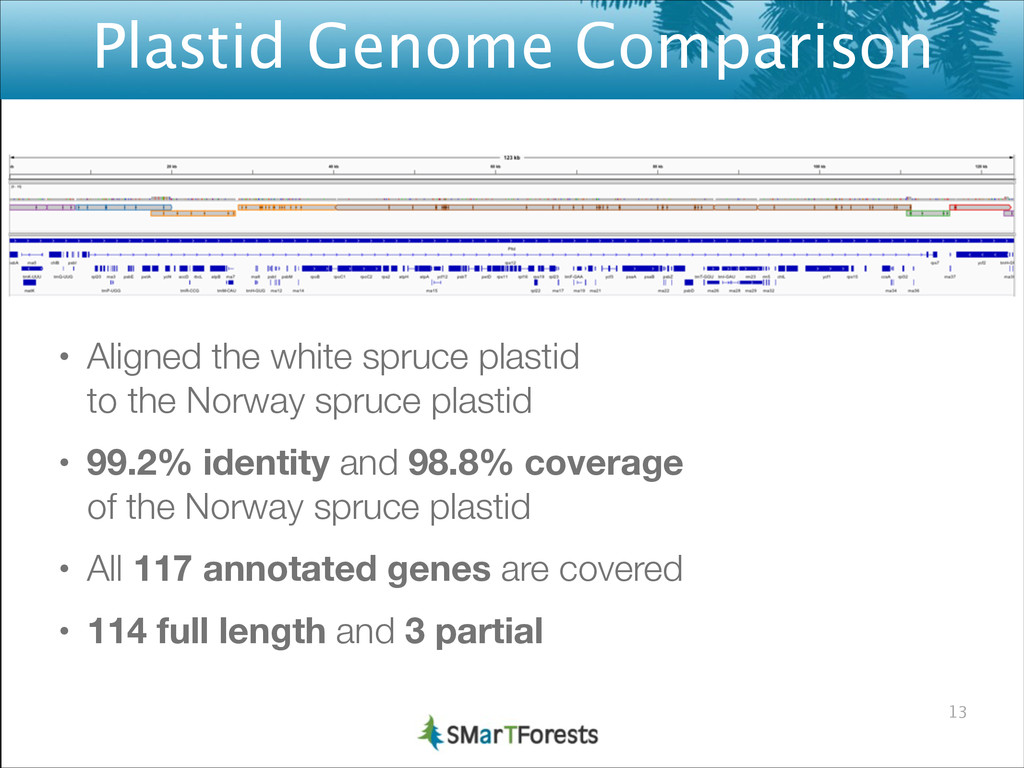{kind=link}
{kind=link}
{kind=link}
{kind=link}
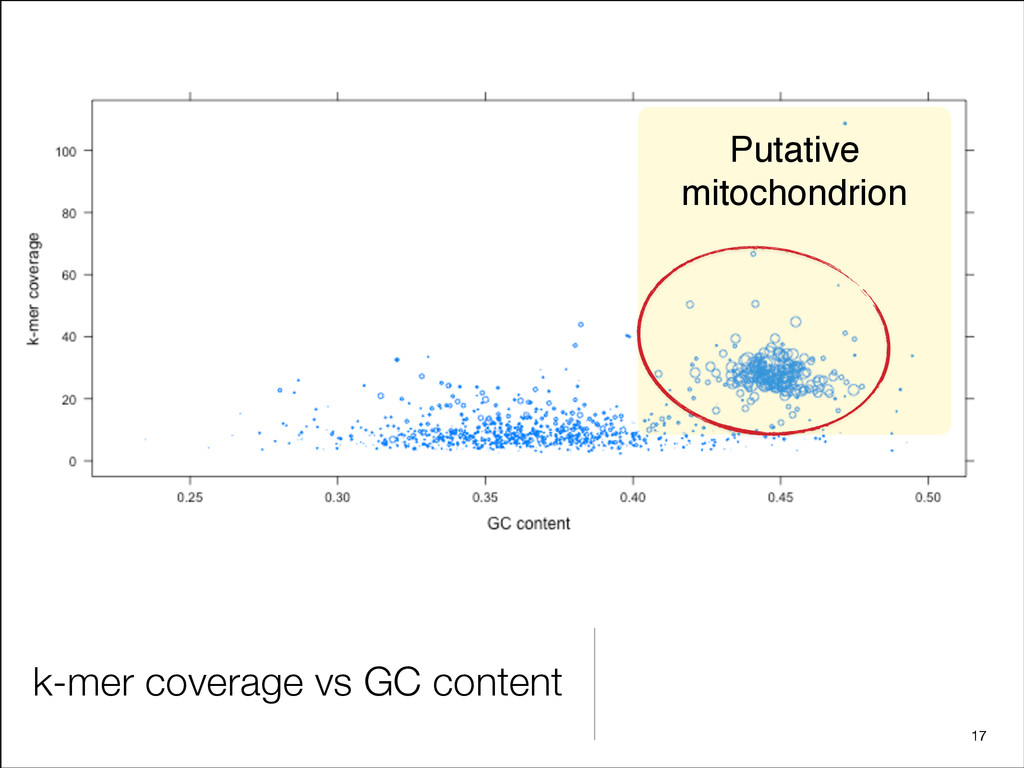{kind=link}
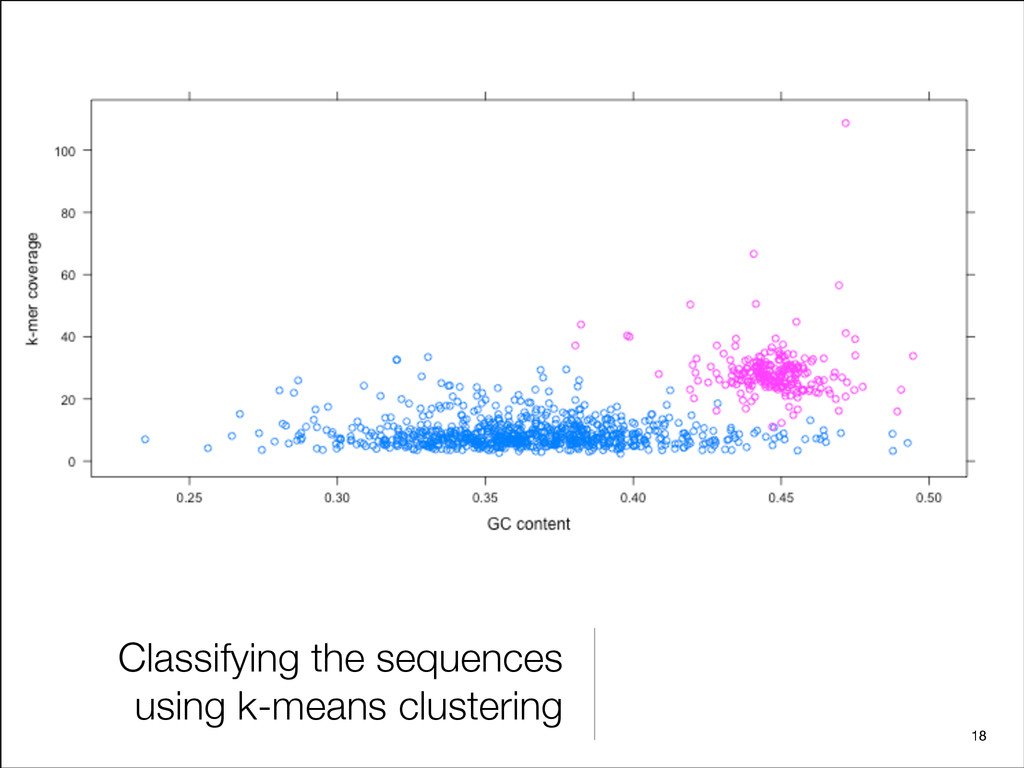{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}