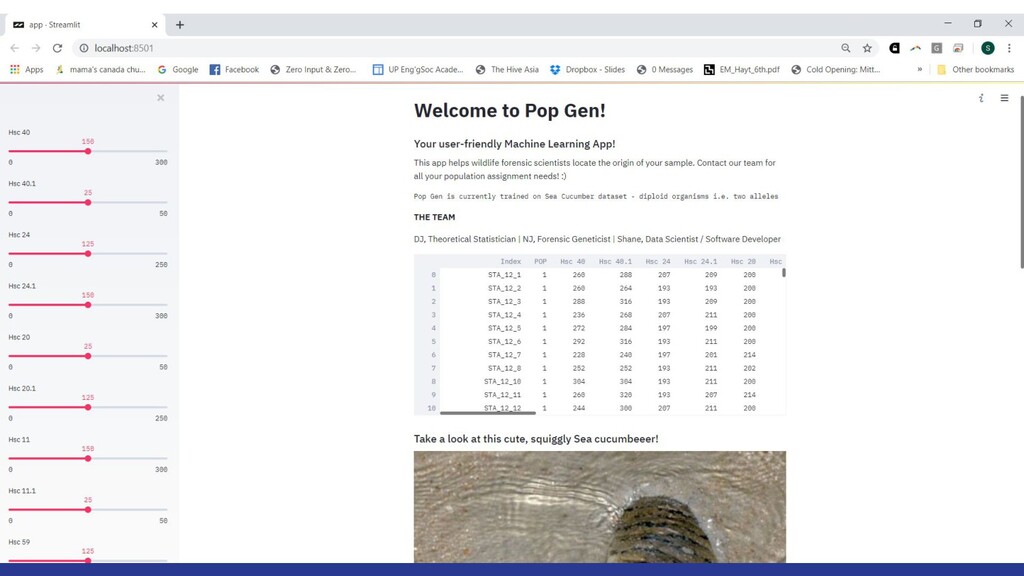
We built an app that could predict the geographic origin of a sea cucumber sample using machine learning. The app could have a huge impact on the mission towards eradicating wildlife exploitation through forensic science.
studied sea cucumbers ◦ Extensive distribution throughout the Indo-Pacific ◦ High economic value: “beche-de-mer” ◦ Can be mass produced in hatcheries • IUCN red list classification: endangered • In the Philippines, trade is regulated by the Department of Agriculture; collecting, trading, or buying of undersized sea cucumbers are prohibited and punishable by law Photo by Ria Tan from singapore [CC BY-SA 2.0 (https://creativecommons.org/licenses/by-sa/2.0)]
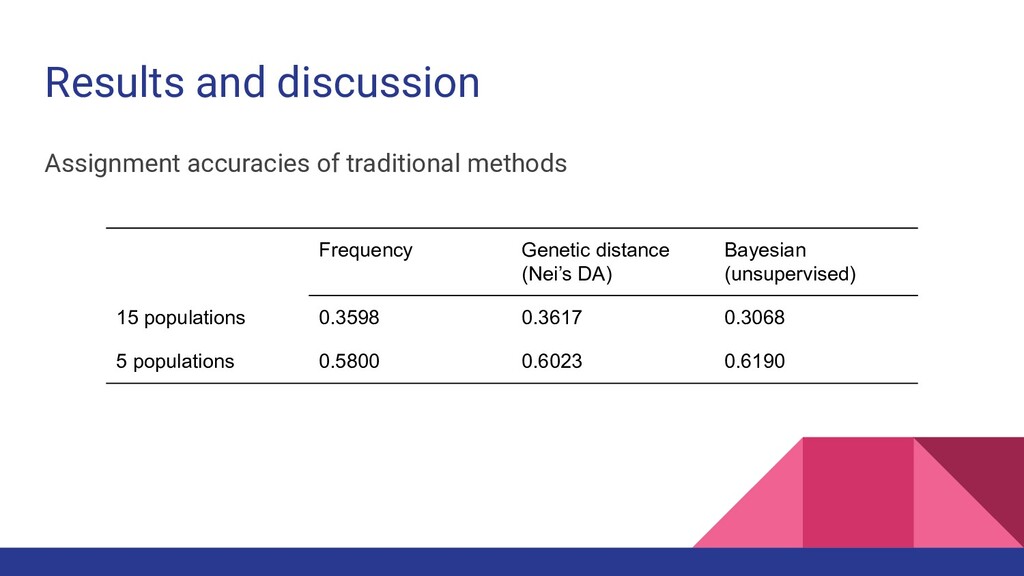
conservation management and wildlife forensics • Answers the investigative question: “Is this sample from a specific population or geographic region?” • Can be used to monitor population specific exploitation • Uses multilocus genotypes (e.g. microsatellite data) • Based on statistical methods ◦ Frequency based - most similar allele frequencies ◦ Genetic distance based - closest genetic distance ◦ Bayesian based - similar patterns of variation (optimizes HWE and loci independence)
i.e. using available genetic information, we will assign individual sandfish into their respective source population ◦ Implications: conservation management, wildlife forensics ◦ Assignment tests may be used to monitor population-specific exploitation Photo by Brian Jones, courtesy of Blue Ventures
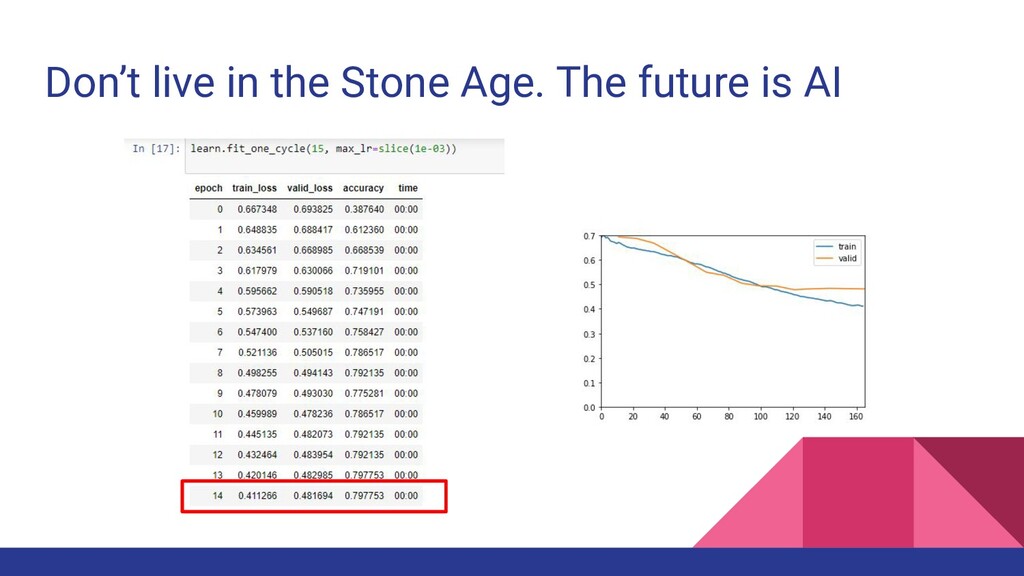
studies have hinted on the use of supervised machine learning in different fields in population genetics (Schrider & Kern, 2018) • Here, we explore if machine learning is applicable in genetic population assignment
samples from 15 sites and genotyped at 13 microsatellite loci Quality control • Samples with 30% missing data were removed from the dataset and excluded from further analysis (528 samples remaining)
frequencies and principal coordinates analysis • Arlequin version 3.5.2.2: F ST calculation and tests for Hardy-Weinberg equilibrium Population genetic assignment • GeneClass2 version 2.0: traditional population assignment using frequency, genetic distance, and Bayesian based methods
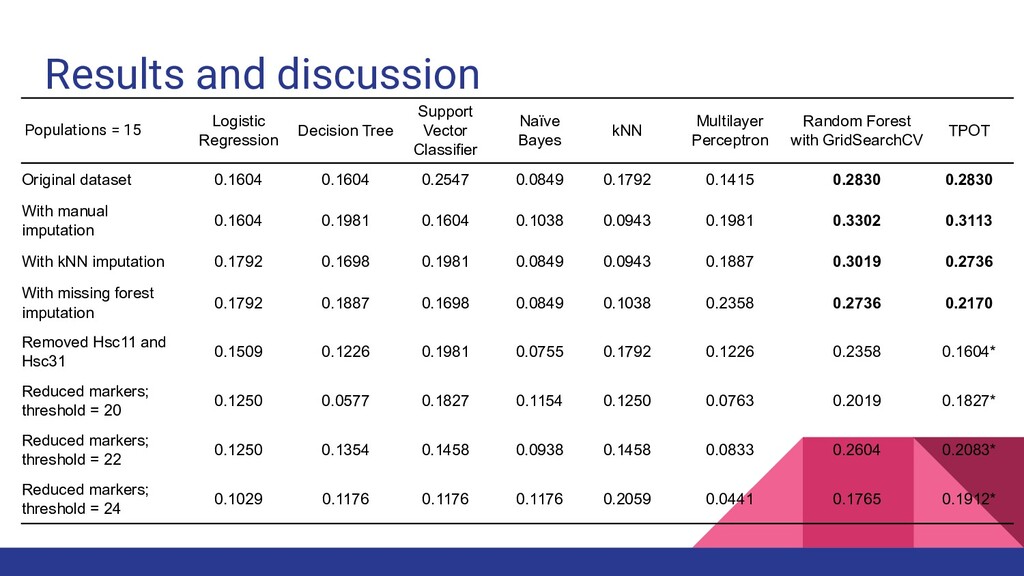
algorithms, missing alleles were imputed using several methods ◦ Manual imputation using MicroDrop version 1.1 ◦ K nearest neighbors (kNN) ◦ Missing forest • In order to check if reducing the loci affects the assignment accuracy, 2 loci (Hsc11 and Hsc31) were removed from analysis • We also reduced the number of samples in order to see if it will affect accuracy
is 0.026 • Populations may be grouped into 5 metapopulations based on population structure Population(s) Group 1 STA, SOR, GUI Group 2 MAS, ROM, CON, TIG, CEB, BOH, DUM Group 3 SAM, GEN, TWI Group 4 COR Group 5 ELN
not perform significantly better from other studies ◦ Larrain et al. (2014): 50% accuracy with F ST = 0.042 • Larrain et al. also highlighted that better results are found in farmed organisms due to reported differences in allele frequencies between farms as a result of artificial selection and less gene flow ◦ Since the sandfish in the study were sampled from natural populations, this may partially explain the low assignment accuracies
was not better than traditional statistical methods at 15 populations and did not differ significantly at 5 populations • Combining related groups together improved the accuracy for both traditional and machine learning methods ◦ Reduced complexity, increased sample size ◦ Increased prior likelihood of assigning samples correctly (from 1/15 to 1/5) • Excluding the traditional methods, the model with highest accuracy was TPOT at 59.43%
conservation management and wildlife forensics • Organisms with very low F ST (less than 0.1) are more challenging to assign • Holothuria scabra in the Philippines have very low global F ST (0.026) which resulted in poor assignment power using both traditional and machine learning models • Reducing the complexity of data resulted in better accuracies • Reducing the number of samples and loci resulted in lower accuracies • Assignment power may be improved by increasing the number of samples, number of loci, or a combination of both
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}