have homology, that is, genes that have been inherited from a common ancestor • Homology is a binary trait; sequences are either homologous or they are not • The concept of homology is often confused with that of sequence similarity (The latter is a continuous trait defined as a percentage of nucleotide positions shared between any two sequence) • Sequence similarity is used to infer homology, but a similarity value can be calculated between any two sequences regardless of their function or evolutionary relationship 4/12/22 Intro to Phylogenies 5
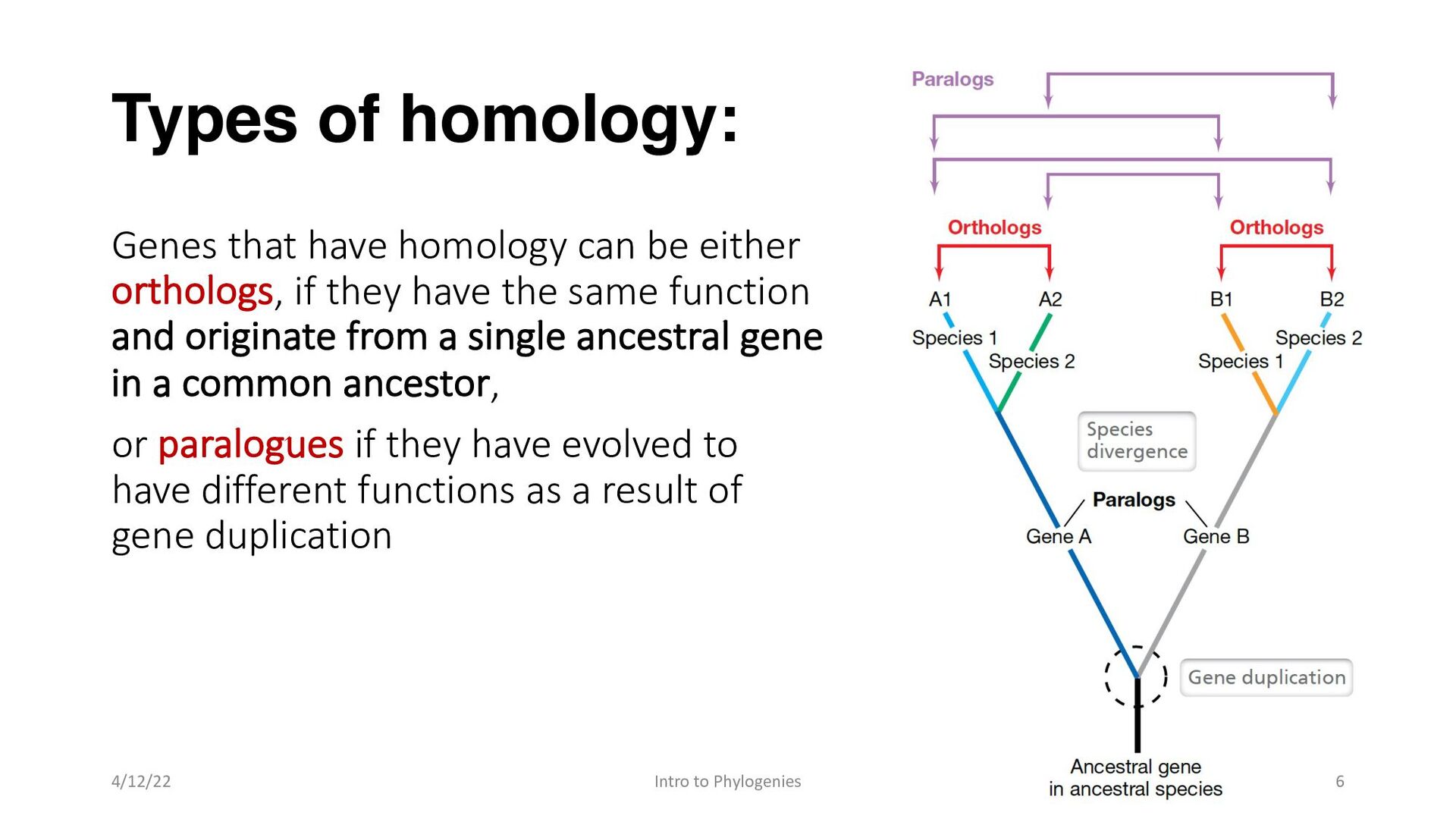
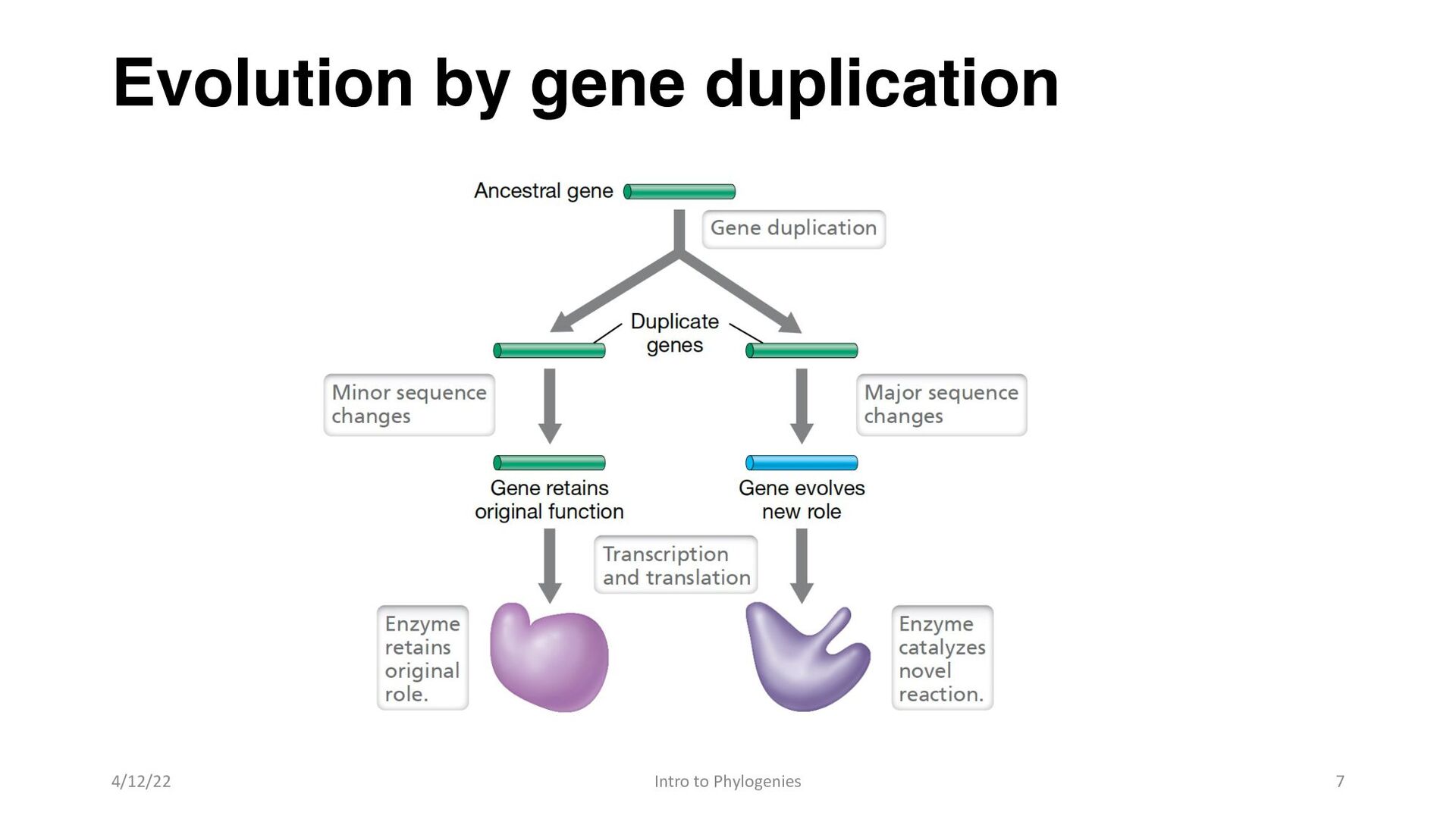
orthologs, if they have the same function and originate from a single ancestral gene in a common ancestor, or paralogues if they have evolved to have different functions as a result of gene duplication 4/12/22 Intro to Phylogenies 6
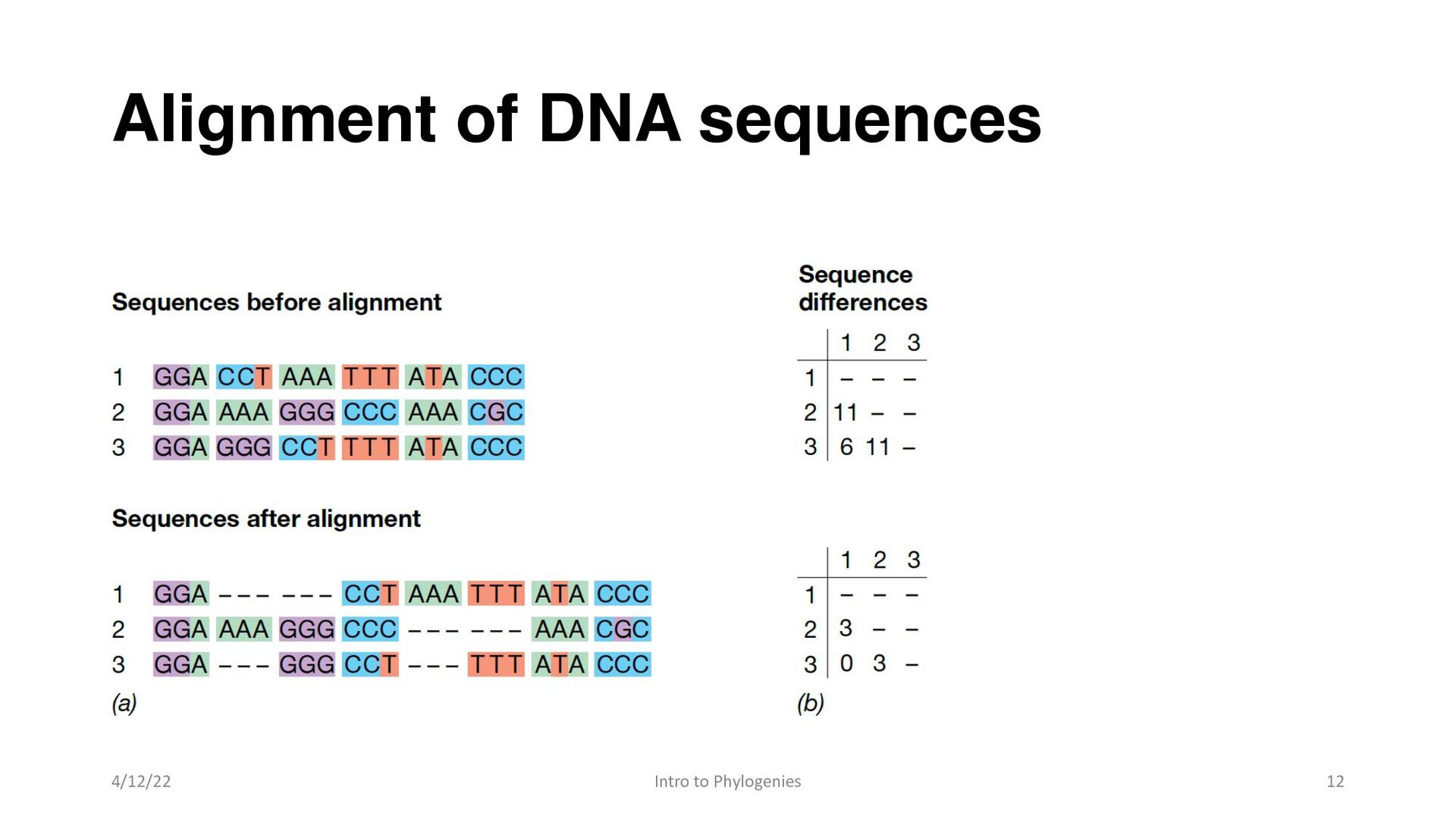
orthologous • genes that have similar function. • Phylogenetic analyses estimate evolutionary changes from the number of sequence differences across a set of homologous nucleotide positions. • Some mutations introduce nucleotide insertions or deletions, and these cause gene sequences to differ in length, making it necessary to align nucleotide positions prior to phylogenetic analysis of gene sequences 4/12/22 Intro to Phylogenies 8
add gaps to molecular sequences in order to establish positional homology, that is, to be sure that each position in the sequence was inherited from a common ancestor of all organisms under consideration • Proper sequence alignment is critical to phylogenetic analysis because the assignment of mismatches and gaps caused by deletions is in effect an explicit hypothesis of how the sequences have diverged from a common ancestral sequence 4/12/22 Intro to Phylogenies 9
a diagram that depicts the evolutionary history of an organism and bears some resemblance to a family tree. • Most microbes have not left fossils and so their ancestors are unknown, but ancestral relationships can be inferred from the DNA sequences of organisms that are alive today • Organisms that share a recent ancestor are likely to share characteristics, and thus phylogenetic trees allow us to make hypotheses about an organism’s characteristics. • Phylogenetic trees are also of great use in taxonomy and species identification 4/12/22 Intro to Phylogenies 10
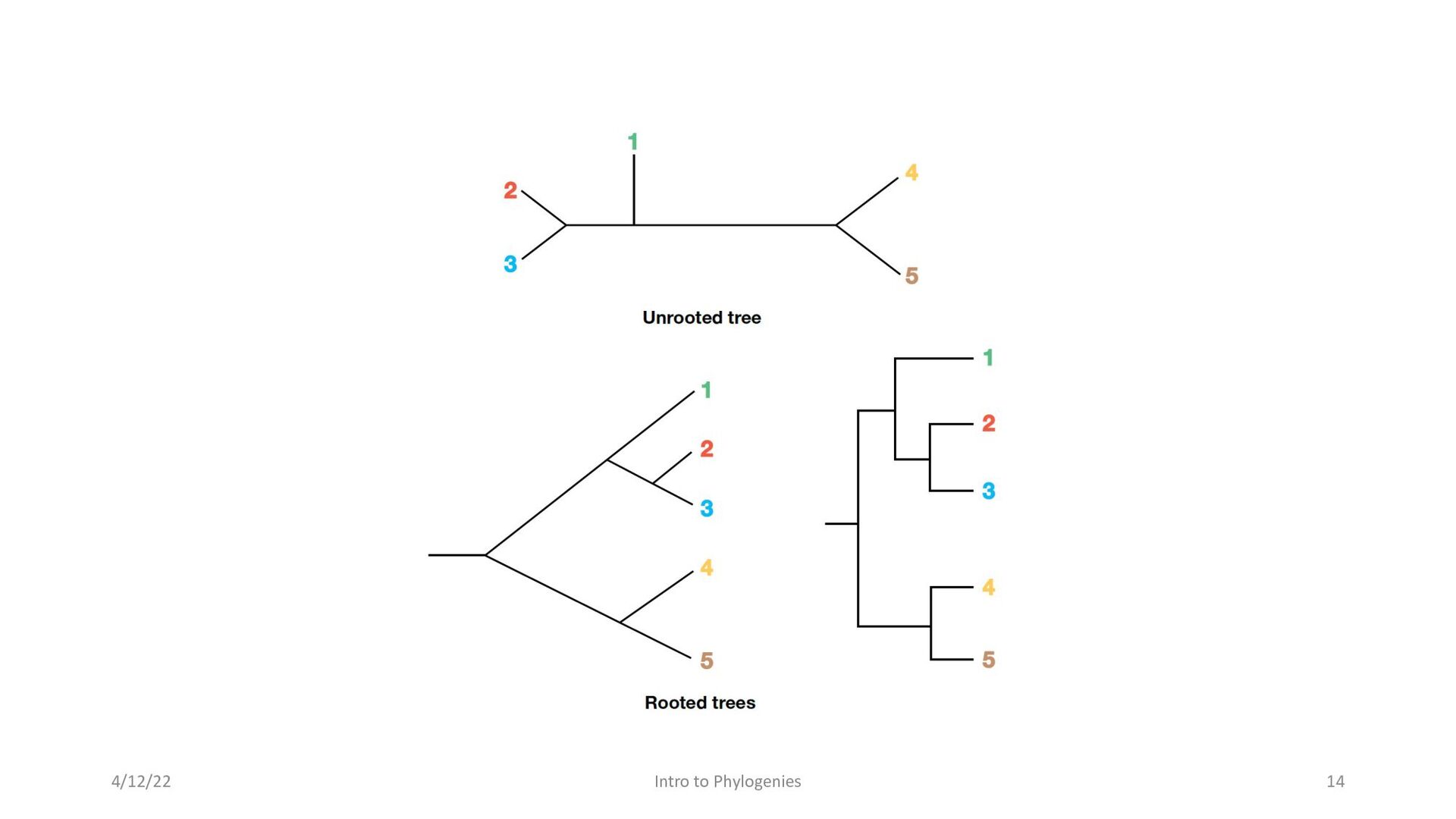
that are either rooted trees or unrooted trees • Rooted trees show the position of the ancestor of all organisms being examined. • Unrooted trees depict he relative relationships among the organisms under study but do not provide evidence of the most ancestral node in the tree. 4/12/22 Intro to Phylogenies 13
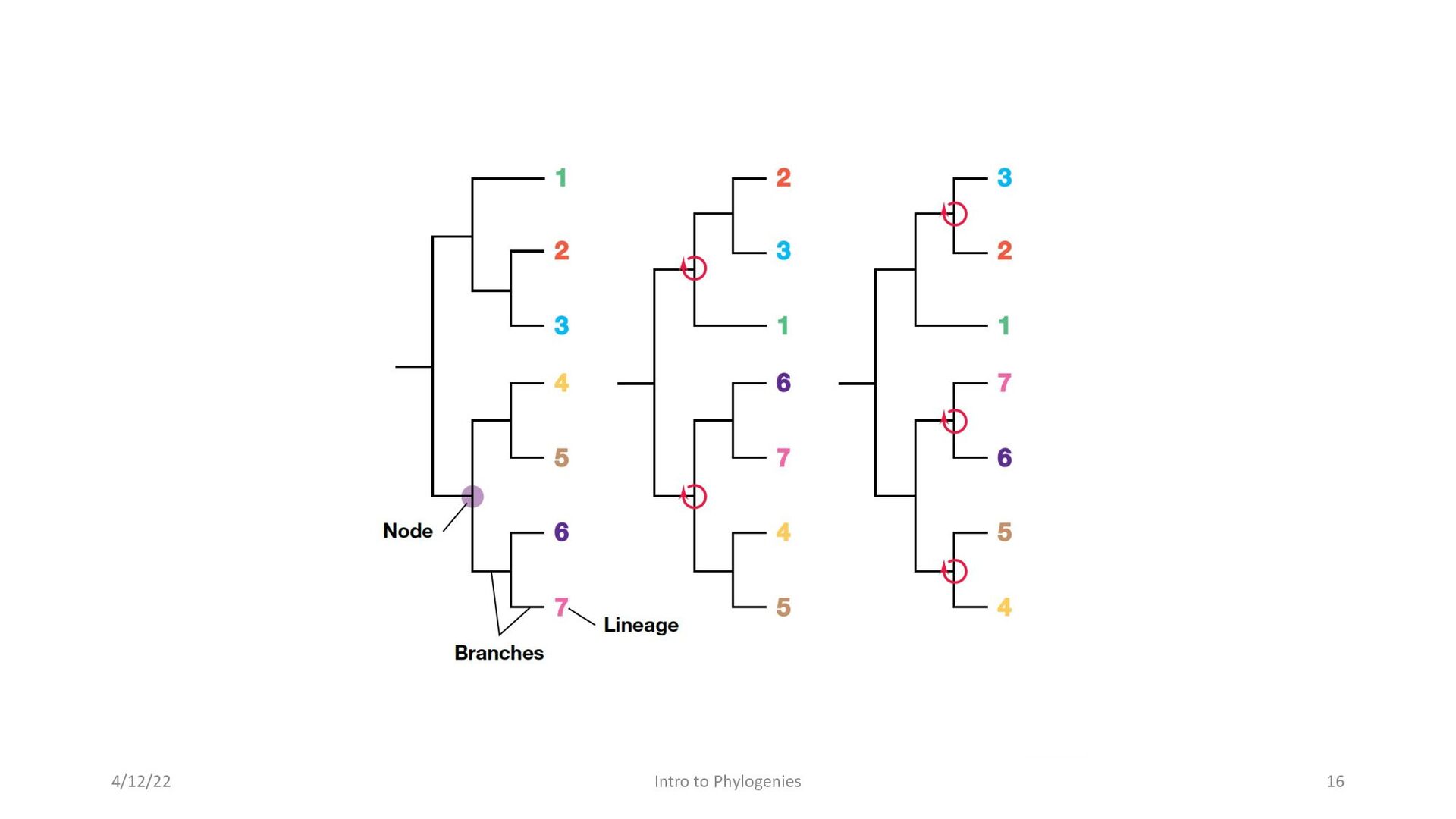
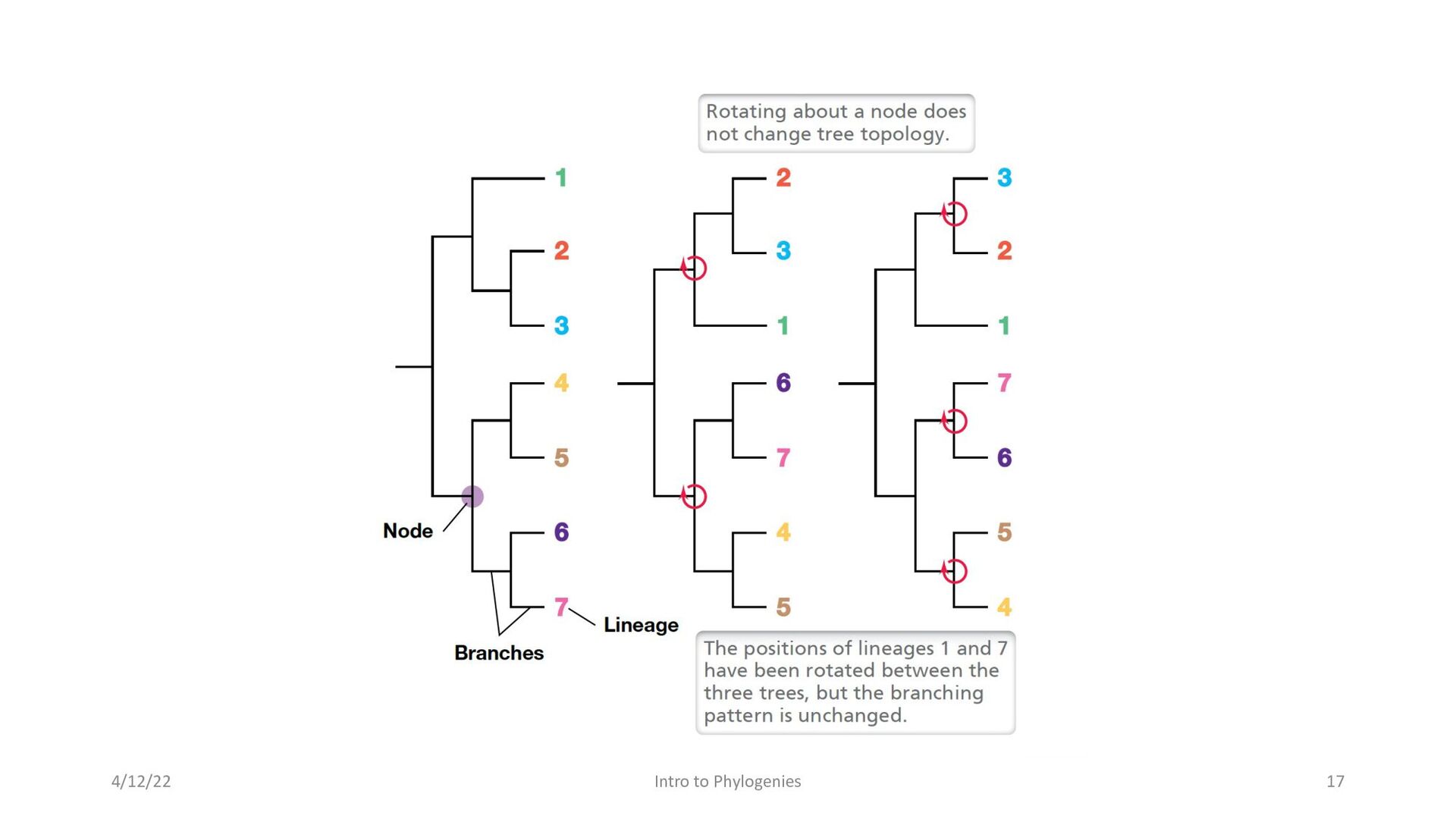
composed of nodes and branches • The tips of the branches in a phylogenetic tree represent species that exist today. • The nodes represent a past stage of evolution where an ancestor diverged into two new lineages. The branch length represents the number of changes that have occurred along that branch. • In a phylogenetic tree, only the position of nodes and the branch lengths are informative; rotation around nodes has no effect on the tree’s topology 4/12/22 Intro to Phylogenies 15
one correct phylogenetic tree that most accurately depicts the evolutionary history of a group of gene sequences, but inferring this tree from sequence data can be a challenging task. • The complexity of the problem is revealed by considering the total number of different trees that can be formed from a random set of sequences 4/12/22 Intro to Phylogenies 18
only three possible trees can be drawn for any four arbitrary sequences. Why? • But if one doubles to eight the number of sequences, now 10,395 trees are possible. (Can you explain this number?) • This complexity continues to expand exponentially such that 2 * 10182 different trees can be drawn to represent 100 arbitrary sequences. • Phylogenetic analysis uses molecular sequence data in an attempt to identify the one correct tree that accurately represents the evolutionary history of a set of sequences 4/12/22 Intro to Phylogenies 19
are available for inferring phylogenetic • trees from molecular sequence data. • The structure of a phylogenetic tree is inferred by applying either an algorithm or some set of optimality criteria. • An algorithm is a programmed series of steps designed to construct a single tree . • Algorithms used to build phylogenetic trees include the unweighted pair group method with arithmetic mean (UPGMA) and the neighbor-joining method. 4/12/22 Intro to Phylogenies 20
parsimony, maximum likelihood, and Bayesian analyses • These methods evaluate many possible trees and select the one tree that has the best optimality score, that is, they select the tree that best fits the sequence data given a discrete model of molecular evolution • Optimality scores are calculated on the basis of evolutionary models that describe how molecular sequences change over time. For example, evolutionary models can account for variation in substitution rates and base frequencies between sequence position 4/12/22 Intro to Phylogenies 21 Methods to infer a phylogenetic tree
into evolutionary history, but it is important to consider the limitations of building and interpreting phylogenetic trees • For example, it can be difficult to choose the true tree based on available sequence data if several different trees fit the data equally well. • Bootstrapping, a statistical method in which information is resampled at random, is an approach used to deal with uncertainty in phylogenetic trees 4/12/22 Intro to Phylogenies 22
that a given node in a phylogenetic tree is supported by the sequence data. • High bootstrap values indicate that a node in the tree is likely to be correct, while low bootstrap values indicate that the placement of a node cannot be accurately determined given the available data. 4/12/22 Intro to Phylogenies 23
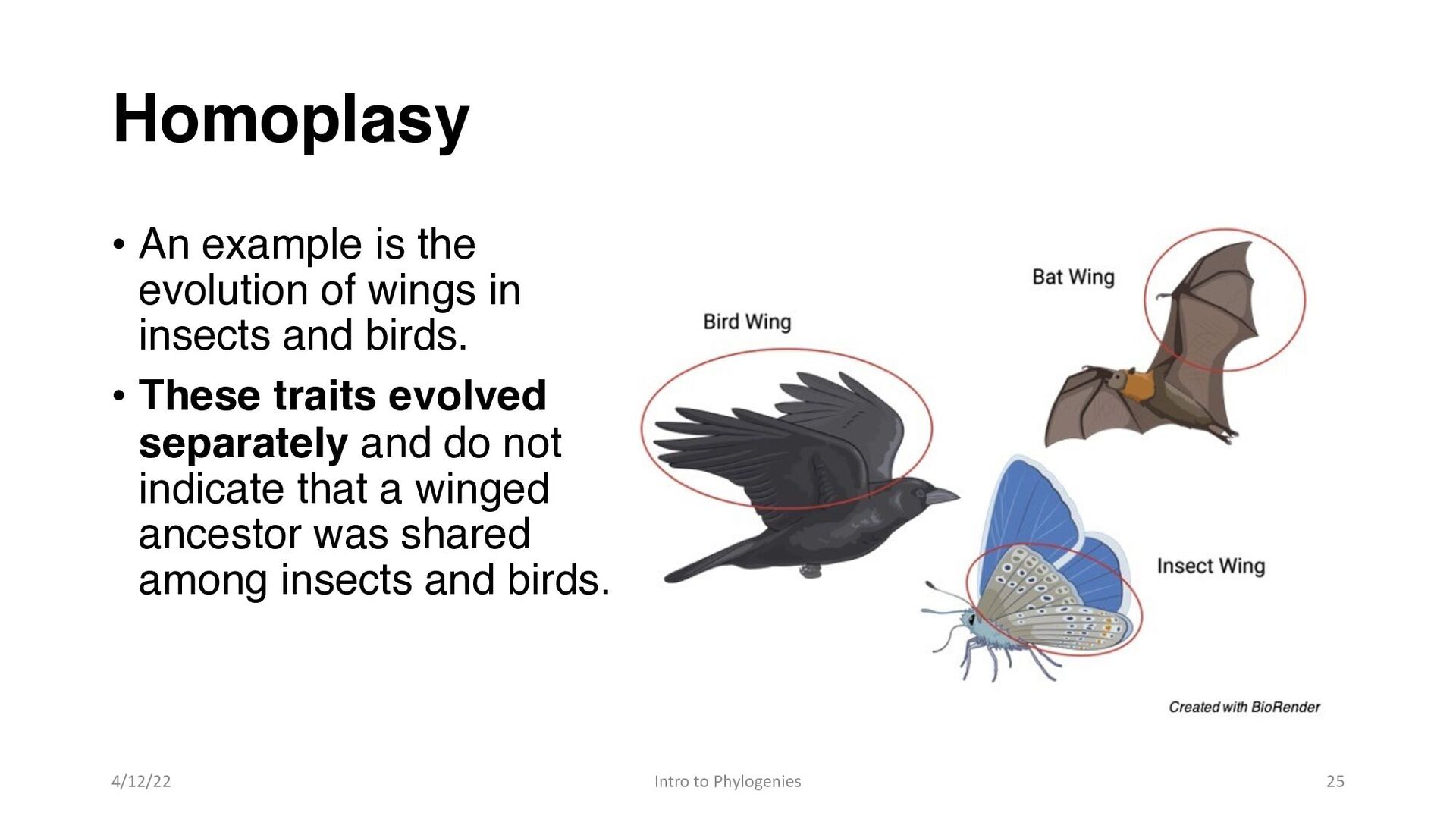
and birds. • These traits evolved separately and do not indicate that a winged ancestor was shared among insects and birds. 4/12/22 Intro to Phylogenies 25 Homoplasy
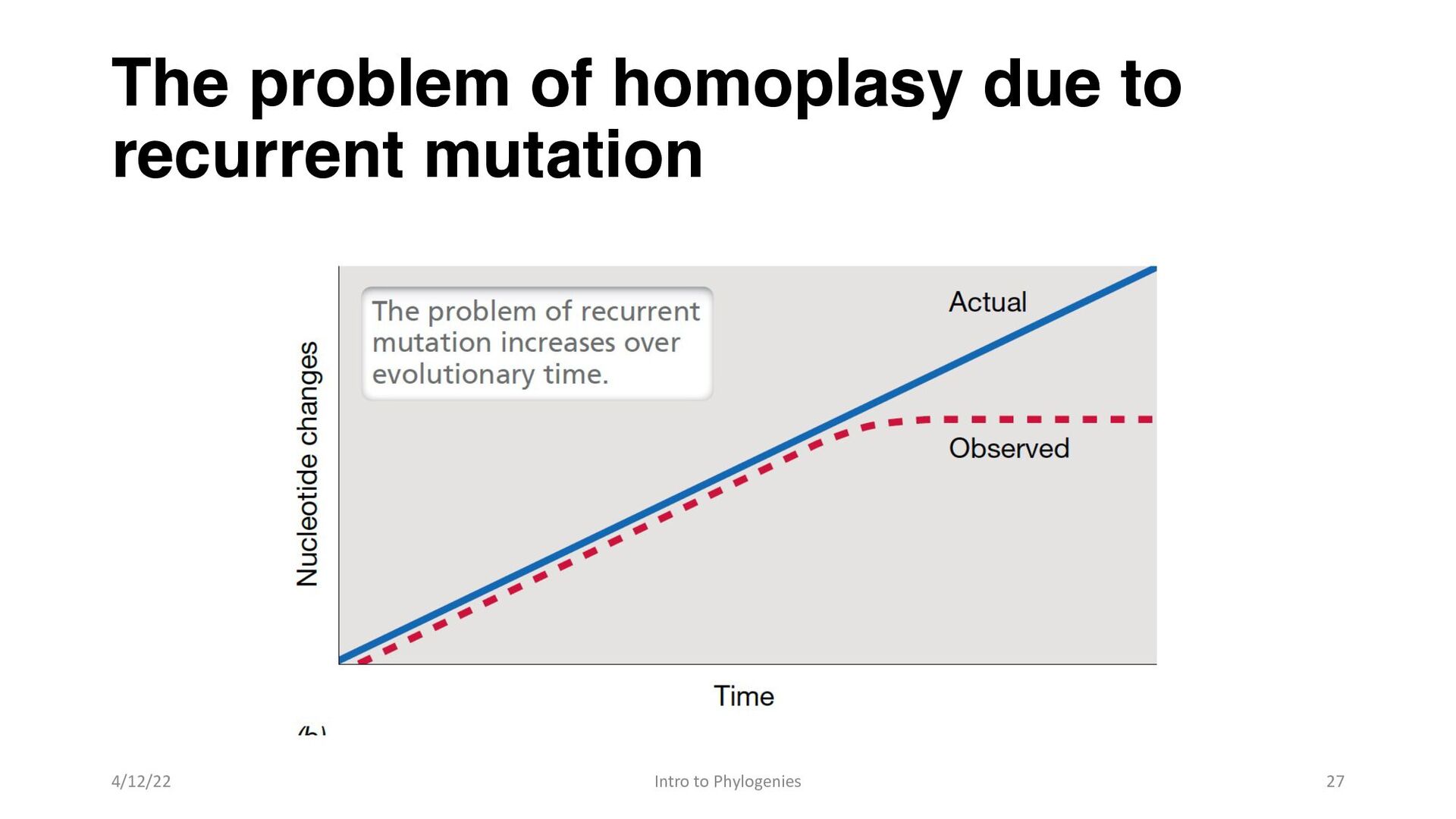
similar sequence positions result from recurrent mutation rather than inheritance from a common ancestor • The problem of homoplasy in molecular phylogeny then increases in proportion to evolutionary time. • As a result of homoplasy, the reconstruction of accurate phylogenetic trees gets more difficult when sequence divergence between organisms is very high 4/12/22 Intro to Phylogenies 26
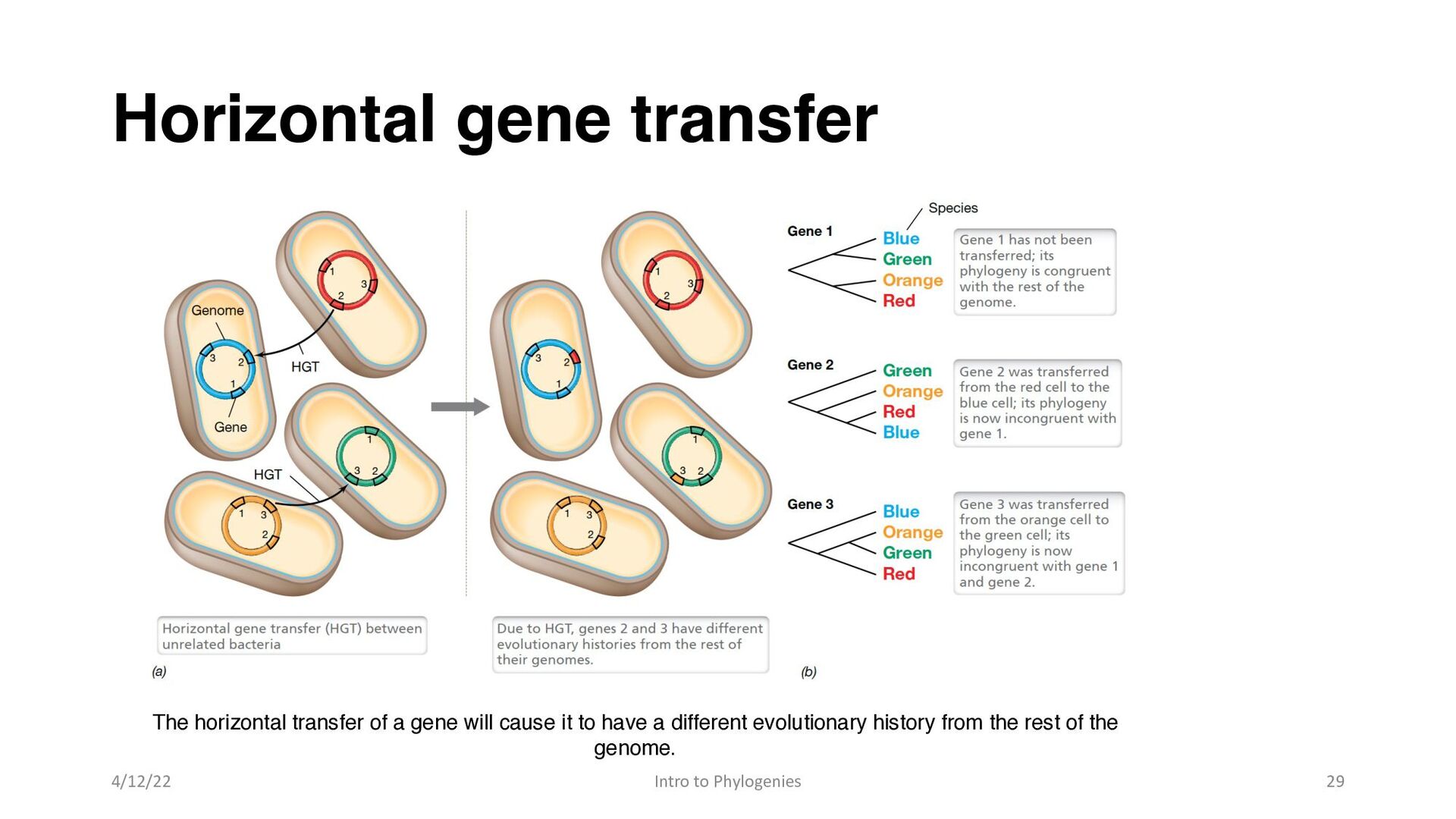
also creates complications when considering the evolutionary history of microorganisms. • When the sequence of a gene is used to infer the phylogeny of an organism, it must be assumed that the gene is inherited in a vertical fashion—from mother to daughter— throughout the evolutionary history of the organism • The horizontal exchange of genes between unrelated organisms violates this assumption. 4/12/22 Intro to Phylogenies 28
between a gene phylogeny, which depicts the evolutionary history of an individual gene, and an organismal phylogeny, which depicts the evolutionary history of the cell • In general, genes encoding SSU rRNAs appear to be transferred horizontally at very low frequencies, and rRNA gene phylogenies agree largely with those prepared from other genes that encode genetic informational functions. • Thus, SSU rRNA gene sequences are generally considered to provide an accurate record of organismal phylogeny. • Nevertheless, many microbial genomes contain genes that have been acquired by horizontal gene transfer at some point in their evolutionary history, and this has important implications for microbial evolution 4/12/22 Intro to Phylogenies 30
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}