Upgrade to Pro
— share decks privately, control downloads, hide ads and more …
Speaker Deck
Features
Speaker Deck
PRO
Sign in
Sign up for free
Search
Search
HiTSeq '16 slides
Search
Sponsored
·
Your Podcast. Everywhere. Effortlessly.
Share. Educate. Inspire. Entertain. You do you. We'll handle the rest.
→
verve
July 08, 2016
Research
2
150
HiTSeq '16 slides
Slides from my HiTSeq '16 talk on Rail-RNA (
http://rail.bio
).
verve
July 08, 2016
Tweet
Share
Other Decks in Research
See All in Research
POI: Proof of Identity
katsyoshi
0
130
Grounding Text Complexity Control in Defined Linguistic Difficulty [Keynote@*SEM2025]
yukiar
0
100
HoliTracer:Holistic Vectorization of Geographic Objects from Large-Size Remote Sensing Imagery
satai
3
610
CoRL2025速報
rpc
4
4.1k
2026年1月の生成AI領域の重要リリース&トピック解説
kajikent
0
180
20年前に50代だった人たちの今
hysmrk
0
140
AIスパコン「さくらONE」の オブザーバビリティ / Observability for AI Supercomputer SAKURAONE
yuukit
2
1.2k
2026-01-30-MandSL-textbook-jp-cos-lod
yegusa
0
150
Thirty Years of Progress in Speech Synthesis: A Personal Perspective on the Past, Present, and Future
ktokuda
0
160
令和最新技術で伝統掲示板を再構築: HonoX で作る型安全なスレッドフロート型掲示板 / かろっく@calloc134 - Hono Conference 2025
calloc134
0
550
自動運転におけるデータ駆動型AIに対する安全性の考え方 / Safety Engineering for Data-Driven AI in Autonomous Driving Systems
ishikawafyu
0
130
2025-11-21-DA-10th-satellite
yegusa
0
110
Featured
See All Featured
Mozcon NYC 2025: Stop Losing SEO Traffic
samtorres
0
140
How to Align SEO within the Product Triangle To Get Buy-In & Support - #RIMC
aleyda
1
1.4k
Neural Spatial Audio Processing for Sound Field Analysis and Control
skoyamalab
0
160
Optimising Largest Contentful Paint
csswizardry
37
3.6k
Claude Code どこまでも/ Claude Code Everywhere
nwiizo
61
52k
Helping Users Find Their Own Way: Creating Modern Search Experiences
danielanewman
31
3.1k
Building an army of robots
kneath
306
46k
Designing for Performance
lara
610
70k
Build your cross-platform service in a week with App Engine
jlugia
234
18k
DBのスキルで生き残る技術 - AI時代におけるテーブル設計の勘所
soudai
PRO
62
49k
B2B Lead Gen: Tactics, Traps & Triumph
marketingsoph
0
53
Six Lessons from altMBA
skipperchong
29
4.1k
Transcript
Scalable analysis of RNA-seq splicing and coverage @AbhiNellore at HiTSeq
‘16 Langmead & Leek Labs Johns Hopkins University http://rail.bio
Alignment …ATACATCAGACTAGACCGTACCACTCATAGACCTAGACCAGATACAG… CAGACTAGACCGTACCACTCATAGACCTAGACCAGATAC chr1 Sometimes, a read correctly aligns to
the reference genome end to end. read
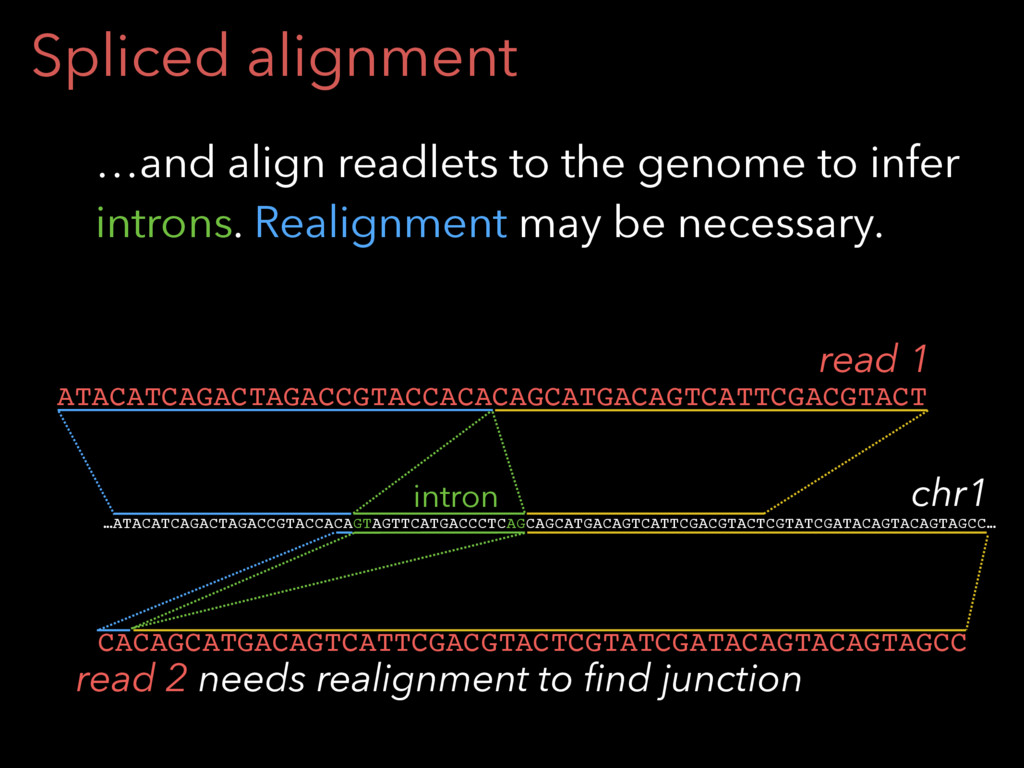
Spliced alignment Other times, exon-exon junctions are overlapped. Rail-RNA divides
the read into readlets… ATACATCAGACTAGACCGTACCACACAGCATGACAGTCATTCGACGTACT ATACATCAGACTAGACCGTACCACA ATCAGACTAGACCGTACCACACAGC GACTAGACCGTACCACACAGCATGA AGACCGTACCACACAGCATGACAGT CGTACCACACAGCATGACAGTCATT CCACACAGCATGACAGTCATTCGAC ACAGCATGACAGTCATTCGACGTAC CAGCATGACAGTCATTCGACGTACT ATACATCAGACTAGA ATACATCAGACTAGAC ATACATCAGACTAGACCG ATACATCAGACTAGACCGT ATACATCAGACTAGACCGTAC ATACATCAGACTAGACCGTACAGC AGCATGACAGTCATTCGACGTACT ATGACAGTCATTCGACGTACT GACAGTCATTCGACGTACT ACAGTCATTCGACGTACT AGTCATTCGACGTACT GTCATTCGACGTACT read readlets
Spliced alignment …ATACATCAGACTAGACCGTACCACAGTAGTTCATGACCCTCAGCAGCATGACAGTCATTCGACGTACTCGTATCGATACAGTACAGTAGCC… intron CACAGCATGACAGTCATTCGACGTACTCGTATCGATACAGTACAGTAGCC ATACATCAGACTAGACCGTACCACACAGCATGACAGTCATTCGACGTACT chr1 read 2 needs
realignment to find junction read 1 …and align readlets to the genome to infer introns. Realignment may be necessary.
Why Rail-RNA • Works on many samples, many cores •
Easy to deploy in different computing environments • Borrows strength across samples • Writes many compact, queryable outputs
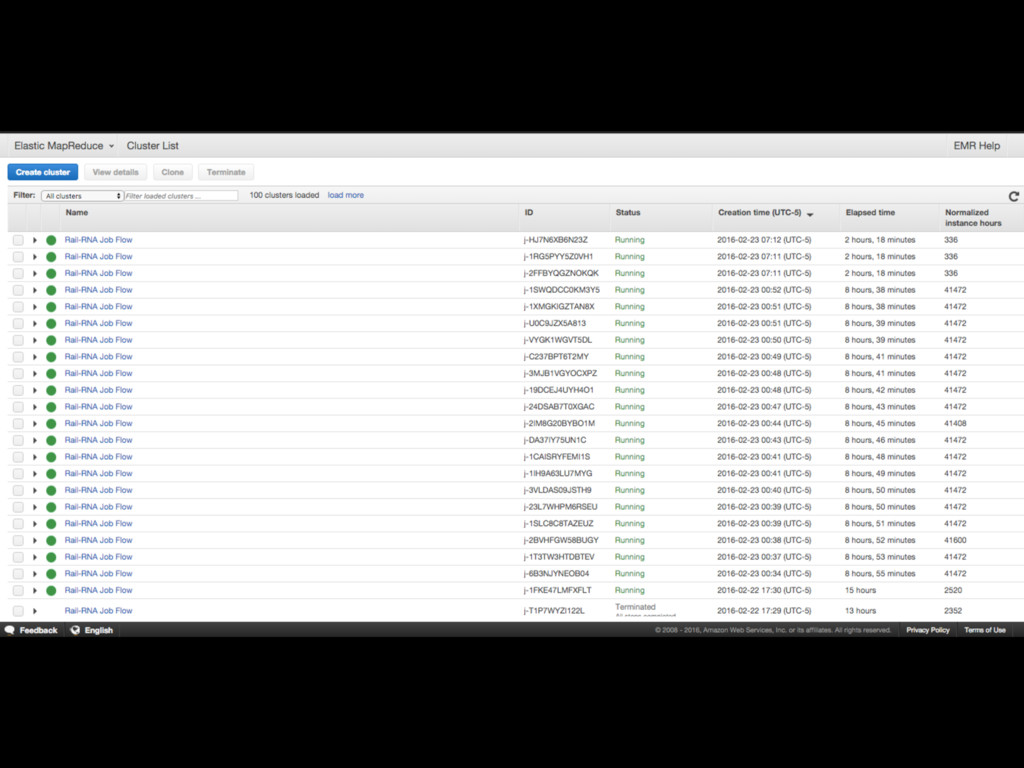
Many samples, many cores
Scaling Use MapReduce. Example: • Divide computer cluster into workers
controlled by a master • Divide problem up into sequence of aggregation and computation steps
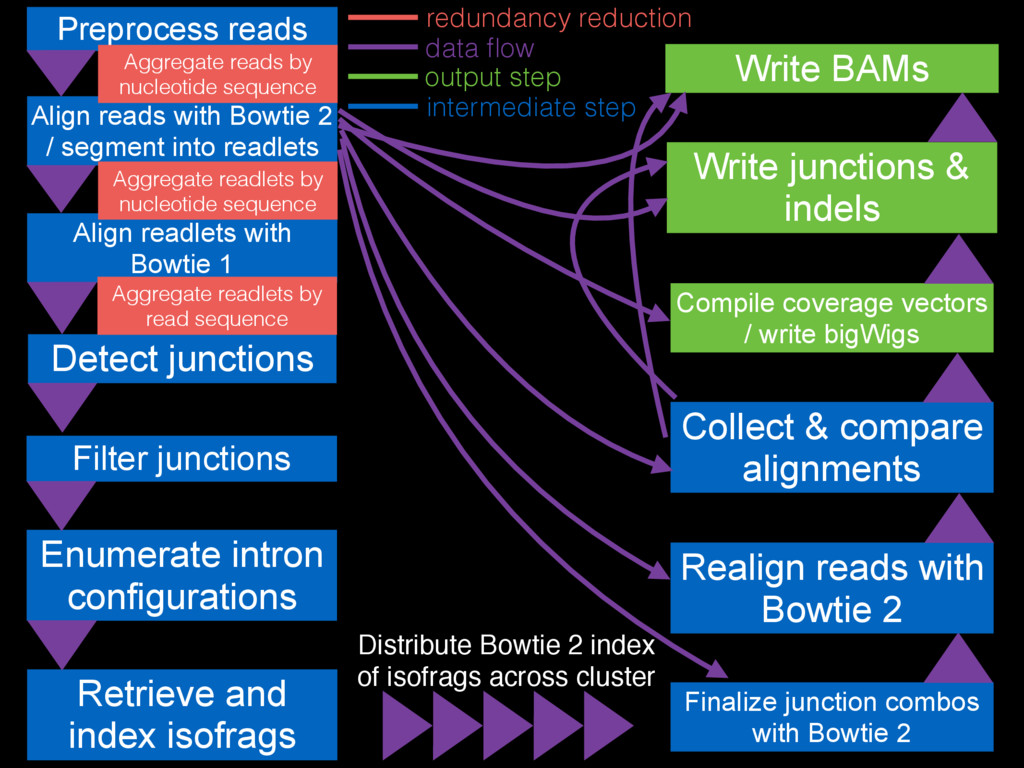
Filter junctions Detect junctions Preprocess reads Align reads with Bowtie
2 / segment into readlets Align readlets with Bowtie 1 Finalize junction combos with Bowtie 2 Enumerate intron configurations Retrieve and index isofrags Realign reads with Bowtie 2 Collect & compare alignments Write BAMs Compile coverage vectors / write bigWigs Write junctions & indels Distribute Bowtie 2 index of isofrags across cluster Aggregate reads by nucleotide sequence Aggregate readlets by nucleotide sequence Aggregate readlets by read sequence data flow redundancy reduction intermediate step output step
Filter junctions Detect junctions Preprocess reads Align reads with Bowtie
2 / segment into readlets Align readlets with Bowtie 1 Finalize junction combos with Bowtie 2 Enumerate intron configurations Retrieve and index isofrags Realign reads with Bowtie 2 Collect & compare alignments Write BAMs Compile coverage vectors / write bigWigs Write junctions & indels Distribute Bowtie 2 index of isofrags across cluster Aggregate reads by nucleotide sequence Aggregate readlets by nucleotide sequence Aggregate readlets by read sequence data flow redundancy reduction intermediate step output step
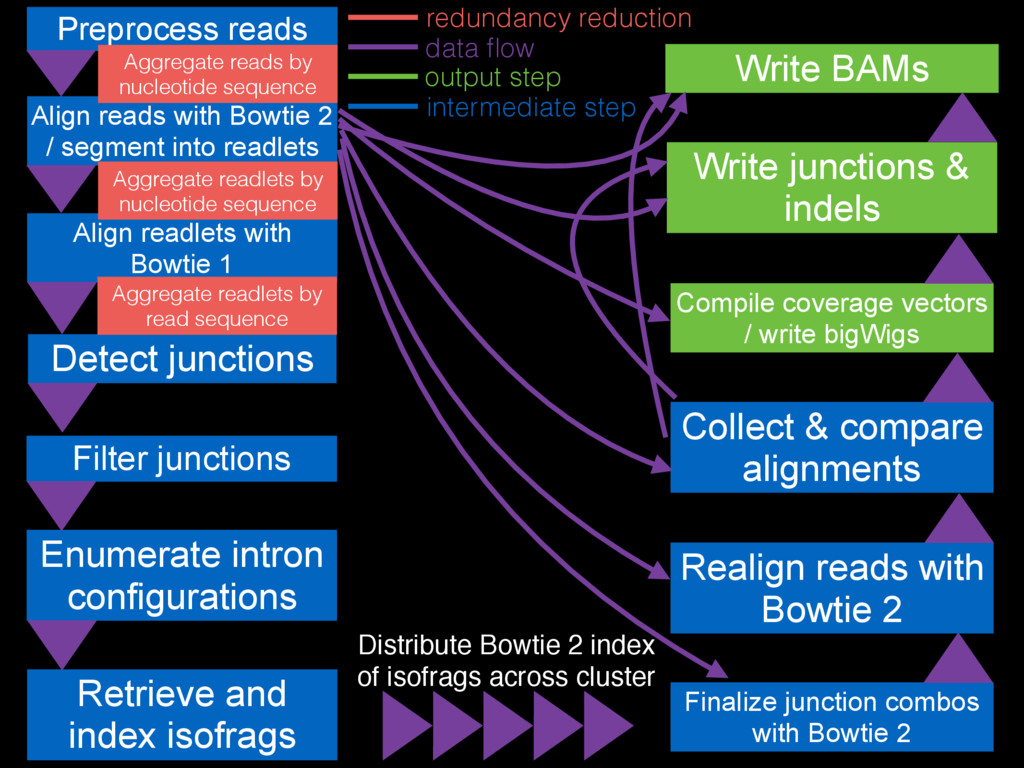
Filter junctions Detect junctions Preprocess reads Align reads with Bowtie
2 / segment into readlets Align readlets with Bowtie 1 Finalize junction combos with Bowtie 2 Enumerate intron configurations Retrieve and index isofrags Realign reads with Bowtie 2 Collect & compare alignments Write BAMs Compile coverage vectors / write bigWigs Write junctions & indels Distribute Bowtie 2 index of isofrags across cluster Aggregate reads by nucleotide sequence Aggregate readlets by nucleotide sequence Aggregate readlets by read sequence data flow redundancy reduction intermediate step output step
Easy to deploy
http://rail.bio rail-rna go elastic —-manifest URLsOf500Samples.txt —-assembly hg38 —-output s3://your-bucket/output_folder
—-core-instance-count 20 —-core-instance-type c3.2xlarge rail-rna go parallel —-manifest URLsOf500Samples.txt —x /path/to/hg38_bowtie_basename —-output /path/to/output_folder Same outputs, different environments, reproducible Cloud w/ AWS EMR Local cluster w/ SGE
Ran Rail-RNA on 49,849 RNA-seq runs from the Sequence Read
Archive (over 150 terabases of reads)
+ • Rapid: 2 weeks to results • Repeatable: http://github.com/nellore/runs
for commands • Inexpensive: ~$1.40/sample
None
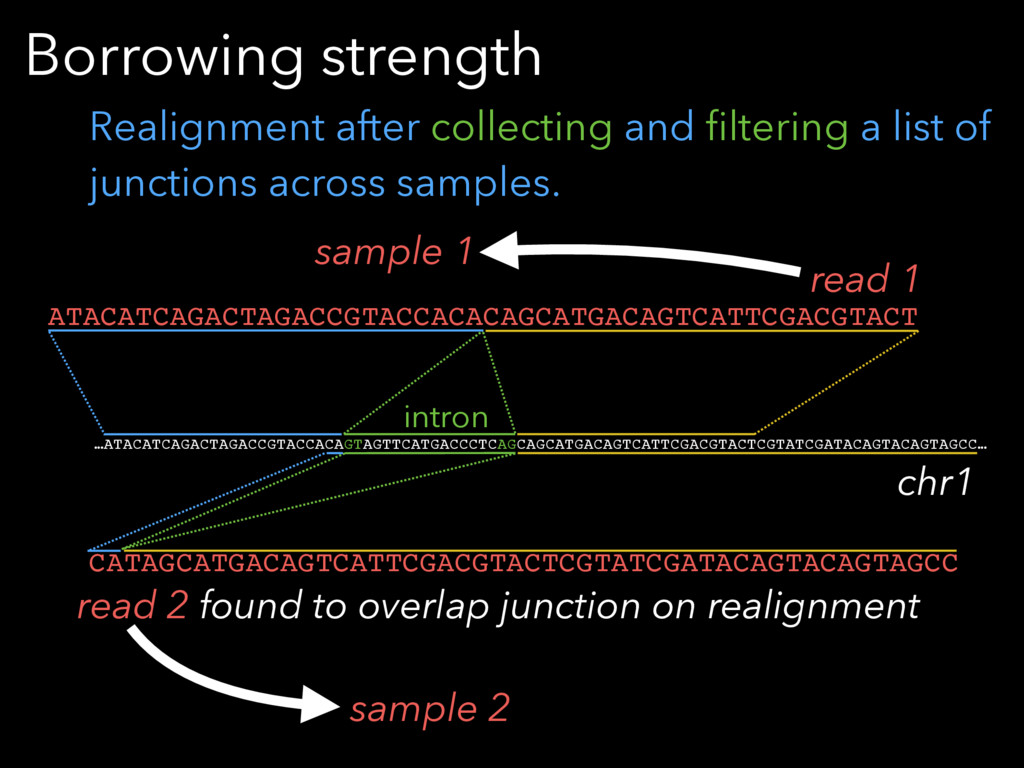
Borrows strength across samples
Borrowing strength …ATACATCAGACTAGACCGTACCACAGTAGTTCATGACCCTCAGCAGCATGACAGTCATTCGACGTACTCGTATCGATACAGTACAGTAGCC… intron CATAGCATGACAGTCATTCGACGTACTCGTATCGATACAGTACAGTAGCC ATACATCAGACTAGACCGTACCACACAGCATGACAGTCATTCGACGTACT chr1 read 2 found
to overlap junction on realignment read 1 Realignment after collecting and filtering a list of junctions across samples. sample 1 sample 2
81,066,376 junctions across 49,849 SRA samples vs. 540,746 annotated junctions
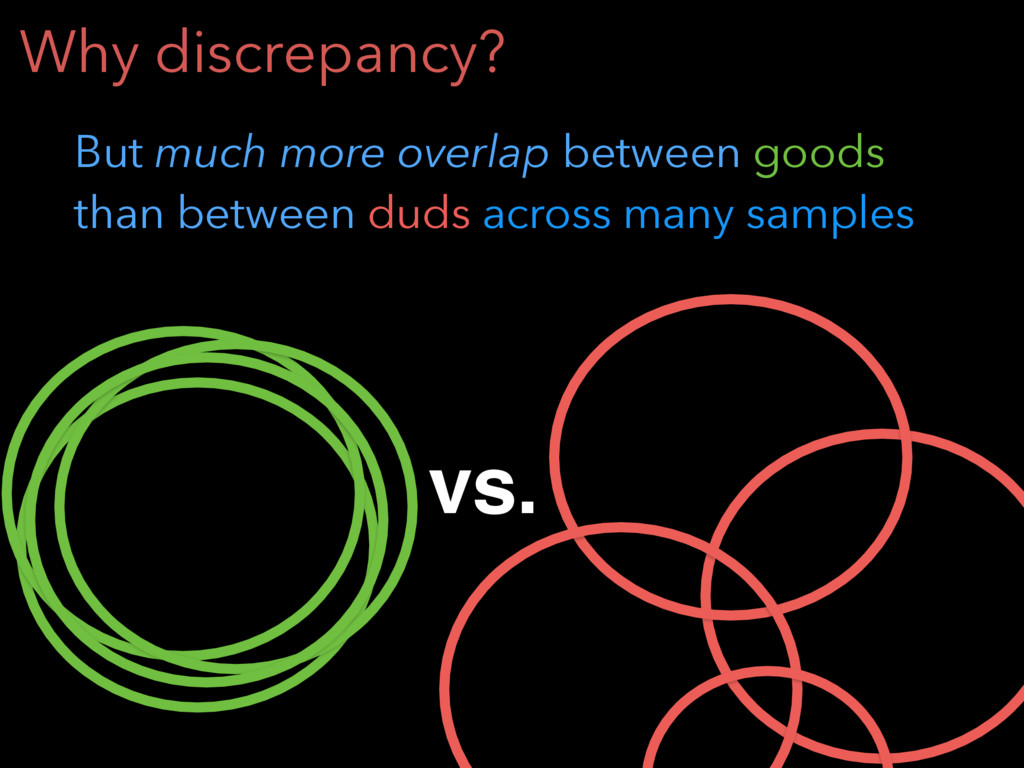
Why discrepancy? On single sample, every aligner finds some good
junctions and some duds goods duds junctions
Why discrepancy? But much more overlap between goods than between
duds across many samples vs.
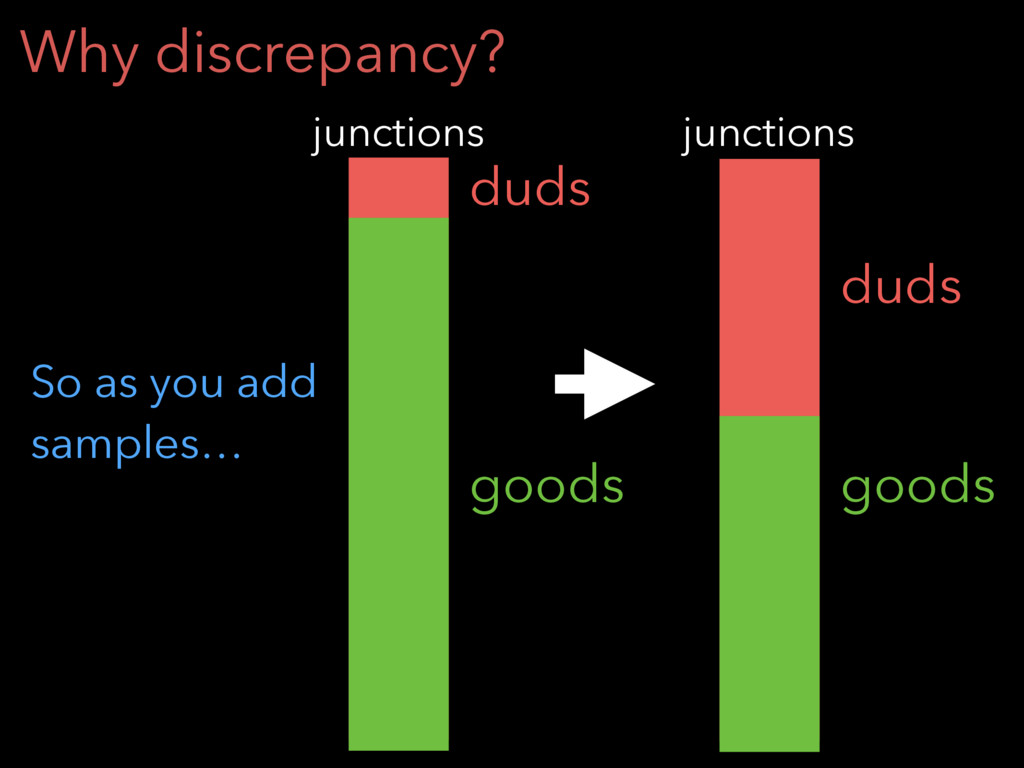
Why discrepancy? So as you add samples… goods duds junctions
goods duds junctions
Junction filter Keep a junction if and only if it’s
initially detected in: (1) 5% of samples OR (2) at least 5 reads in any one sample
Rail-RNA: accuracy (mean ± stdev) exon-exon junction accuracy metrics across
20 GEUVADIS-based simulations Precisions Recalls F-scores Rail single .984 ± .000 .880 ± .004 .929 ± .002 Rail all no filter .846 ± .002 .957 ± .001 .898 ± .001 Rail all filter .976 ± .000 .939 ± .003 .957 ± .002
Writes compact outputs
Compact outputs • junction X sample table • 17 GB
compressed for 50k SRA samples • v1 spans 21.5k samples: available at http://intropolis.rail.bio • v2 w/ 50k coming • coverage bigWigs • 10x smaller than BAM
Annotation-agnostic pipeline derfinder Leo Collado-Torres Alyssa Frazee http://rail.bio biocLite("derfinder") sidesteps
assembly & annotation limitations resolves isoform-level features
http://docs.rail.bio
https://github.com/nellore/rail tested!
Rail-RNA: Scalable analysis of RNA-seq splicing and coverage http://rail.bio Ben
Langmead Jeff Leek Leo Collado-Torres Andrew Jaffe José Alquicira Hernández Summer intern: Jamie Morton Chris Wilks Jacob Pritt
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}