papers) with the introduc:on of next-‐genera:on sequencers • Allowed to analyze en:re gene expression programs • In principle, any high-‐ throughput sequencing technology can be used • Bioinforma:cs tools for RNA-‐seq ~2009 (e.g. TopHat)
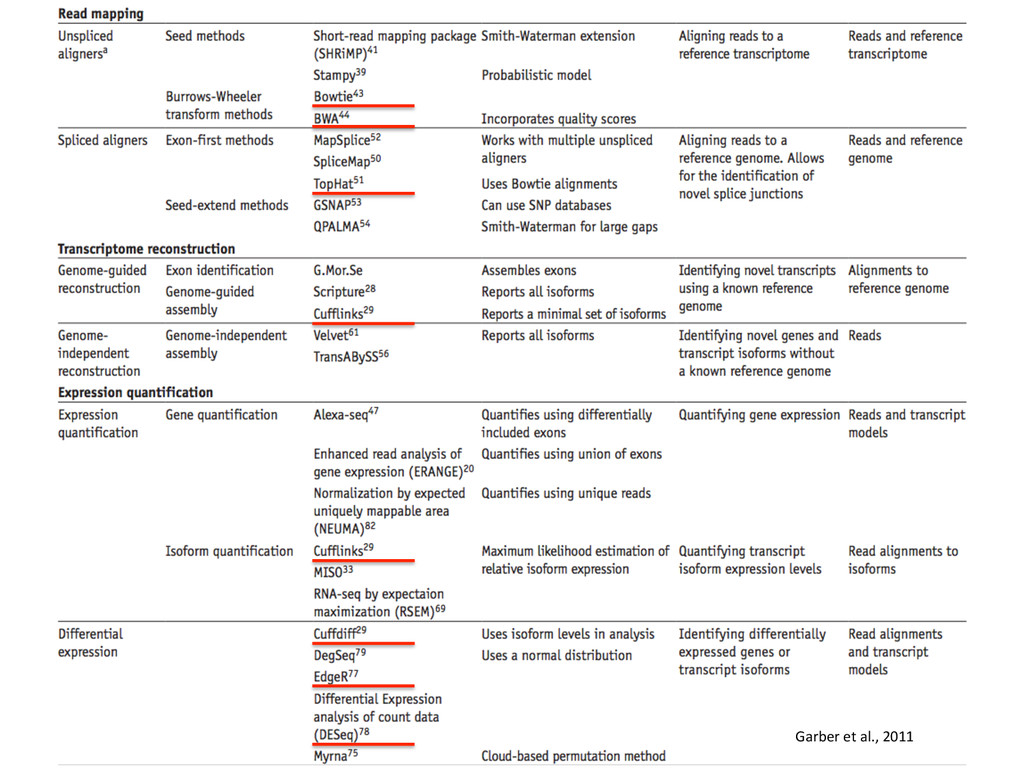
novo assembly (De Bruijn graphs) – Genome unknown or of poor quality 2. Genome alignment – Genome available – Transcriptome unknown or of poor quality – Allows finding new splice junc:ons, polya cleavage sites, etc. 3. Transcriptome alignment – Genome available – Comprehensive transcriptome available
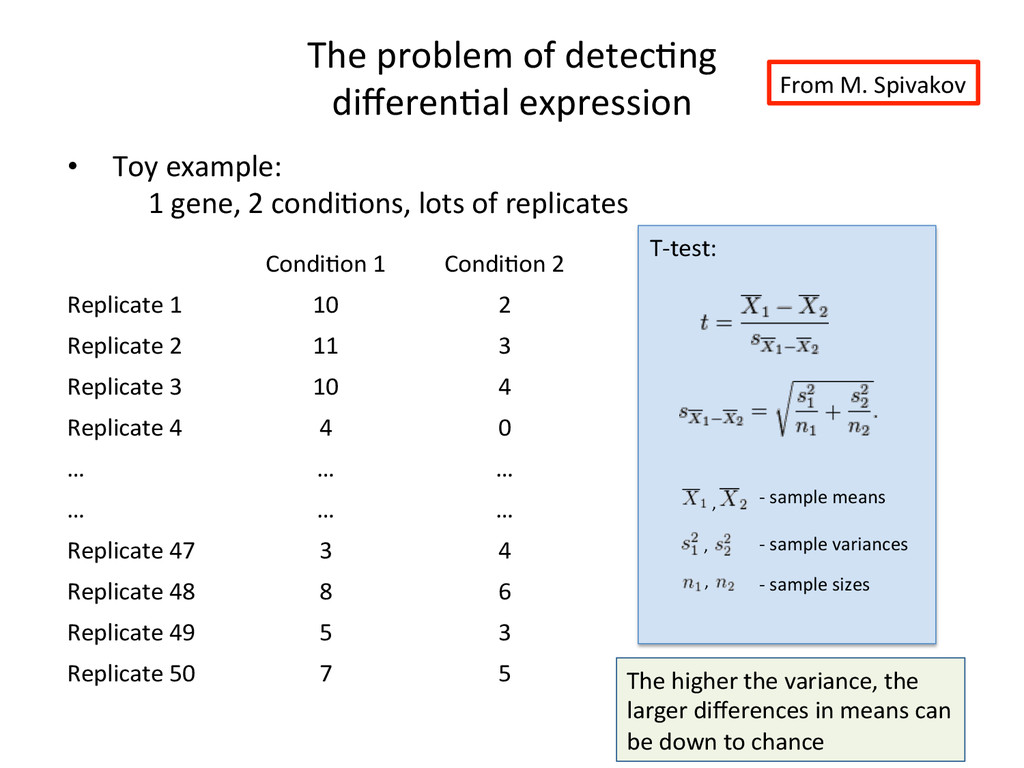
Toy example: 1 gene, 2 condi:ons, lots of replicates • When the number of replicates is very small: – Can’t robustly es:mate popula&on variance from sample variance – Can’t assume normal distribu:on for count data T-‐test: , , -‐ sample variances -‐ sample means , -‐ sample sizes The higher the variance, the larger differences in means can be down to chance This is why more sophis:cated tools are needed From M. Spivakov
Limita:ons on cDNA synthesis and library prepara:on • Challenges in current mapping algorithms Future: • Further development of third(fourth)-‐genera:on sequencing: – Higher detec:on quality – Longer read length • Single cell RNA-‐seq Schadt et al., 2010 Ozsolak et al., 2011
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
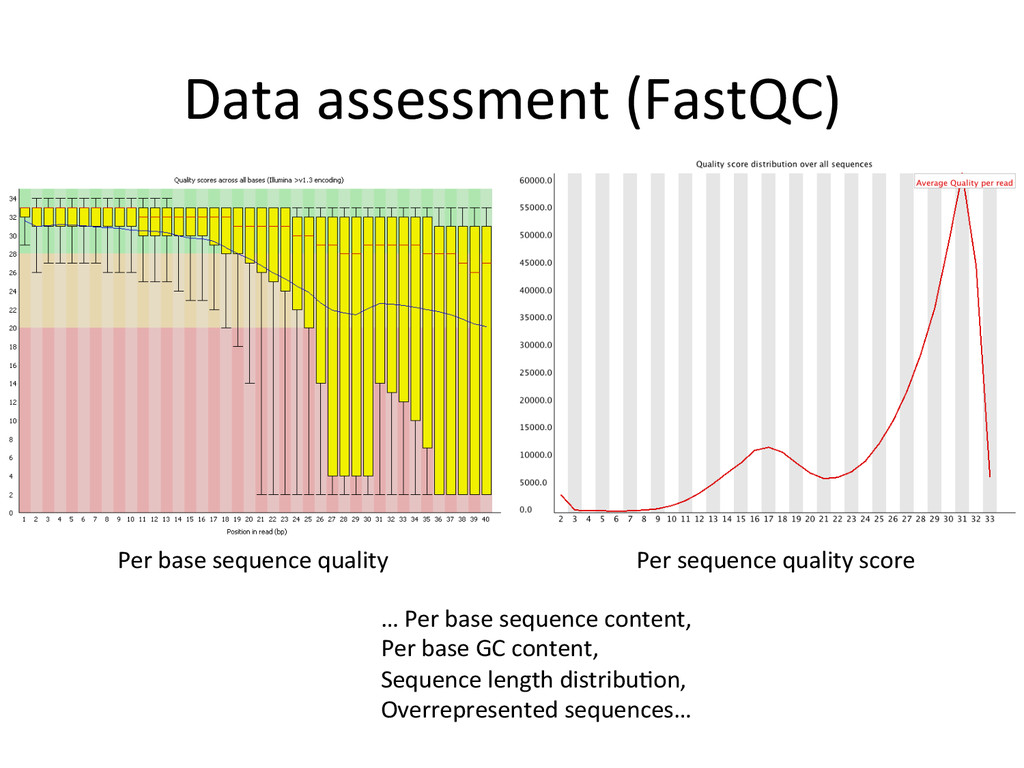{kind=link}
{kind=link}
{kind=link}
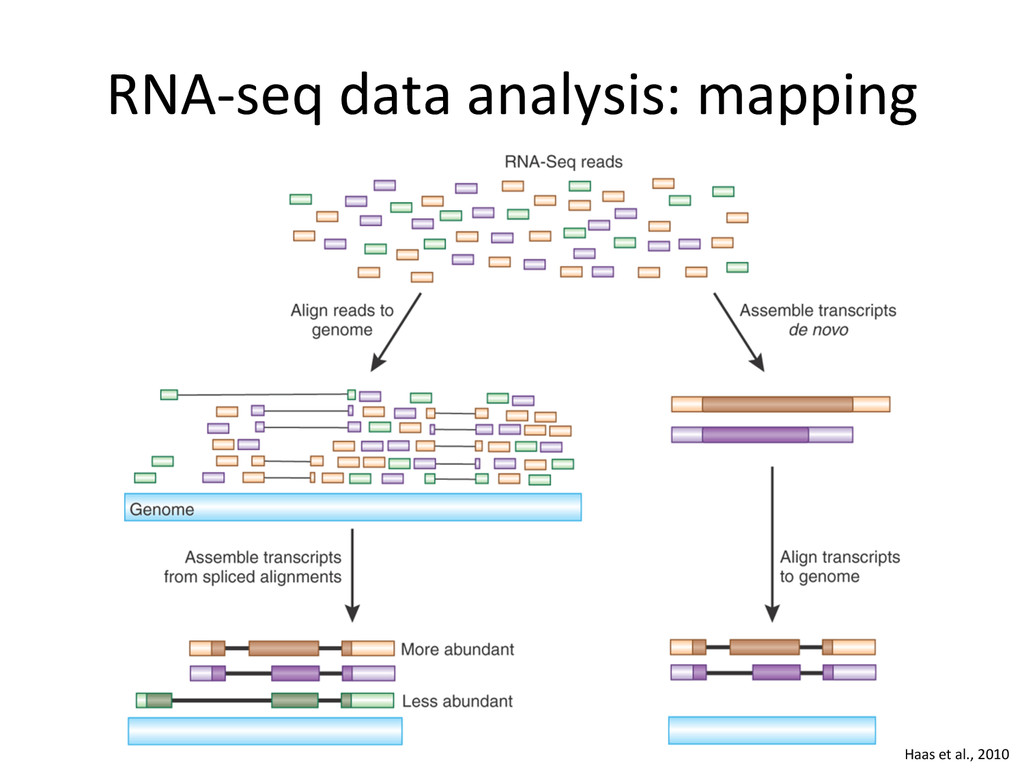{kind=link}
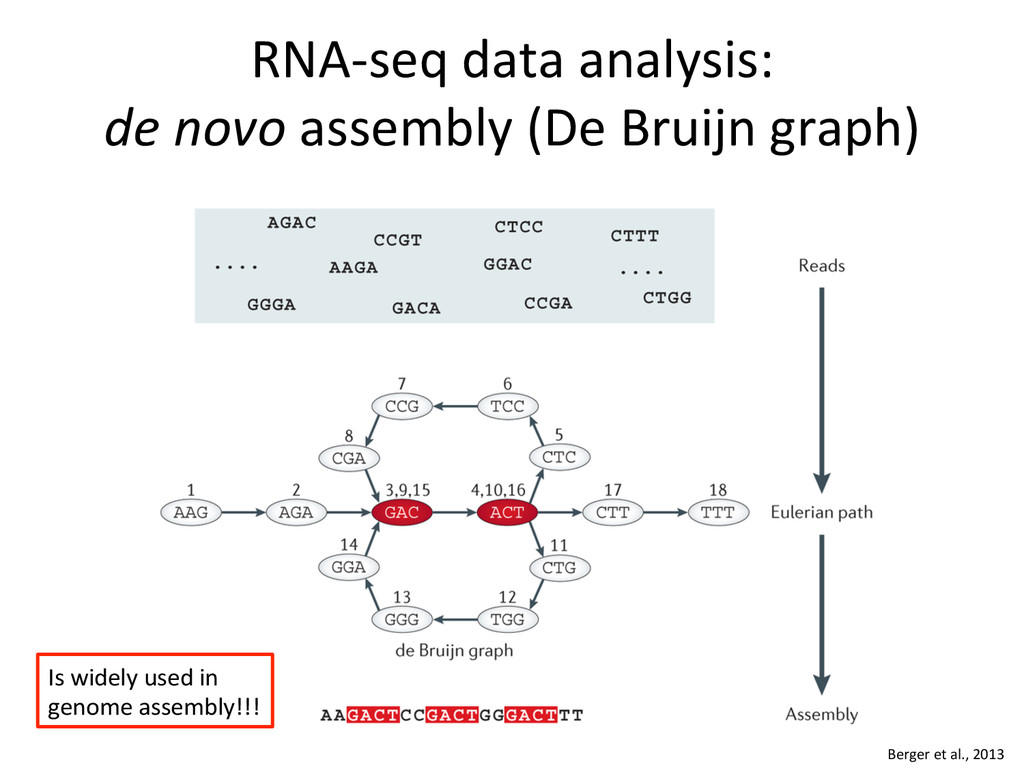{kind=link}
{kind=link}
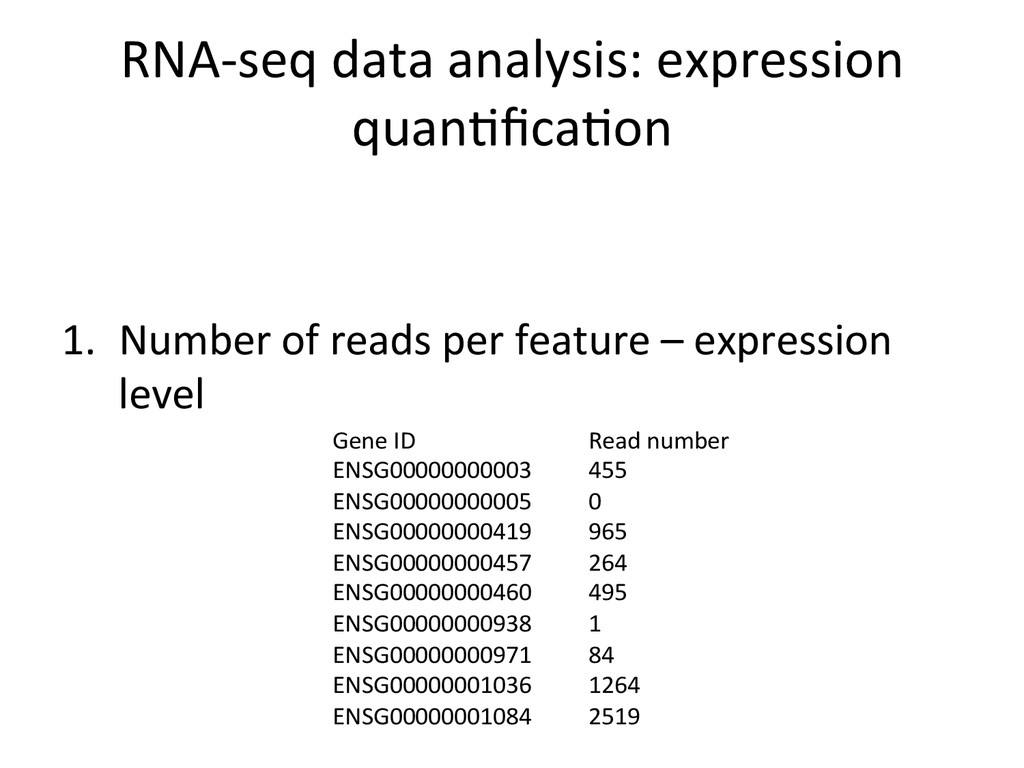{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}