support for Cellular Genetics programme (~180 people) • We work on both upstream (processing raw data) and downstream (gaining biological insights) analysis pipelines • We interact with all of the faculty members in the programme • We move programme’s pipelines to the Cloud (Sanger introduced Flexible Compute Environment ~1.5 years ago)
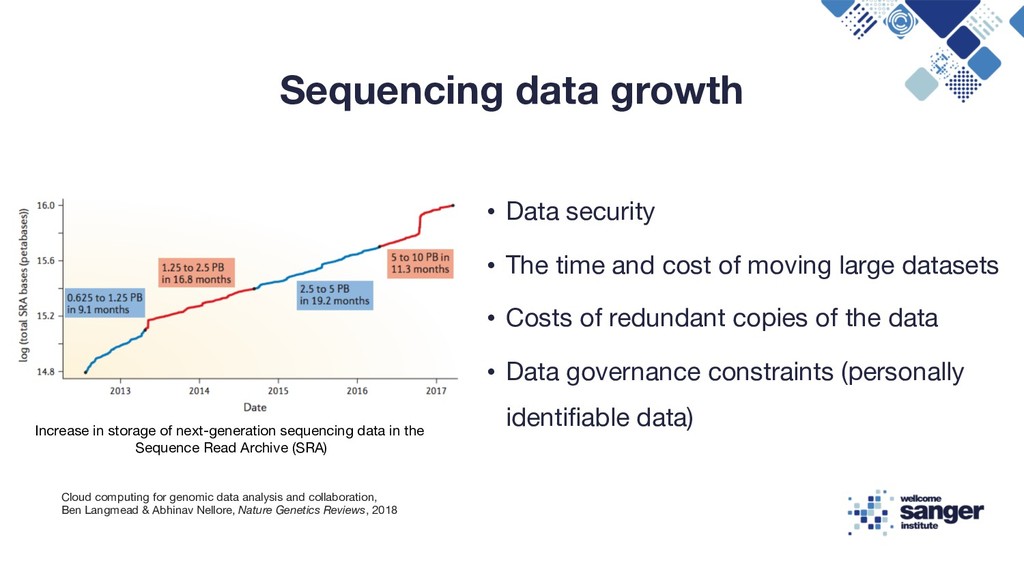
cost of moving large datasets • Costs of redundant copies of the data • Data governance constraints (personally identifiable data) Increase in storage of next-generation sequencing data in the Sequence Read Archive (SRA) Cloud computing for genomic data analysis and collaboration, Ben Langmead & Abhinav Nellore, Nature Genetics Reviews, 2018
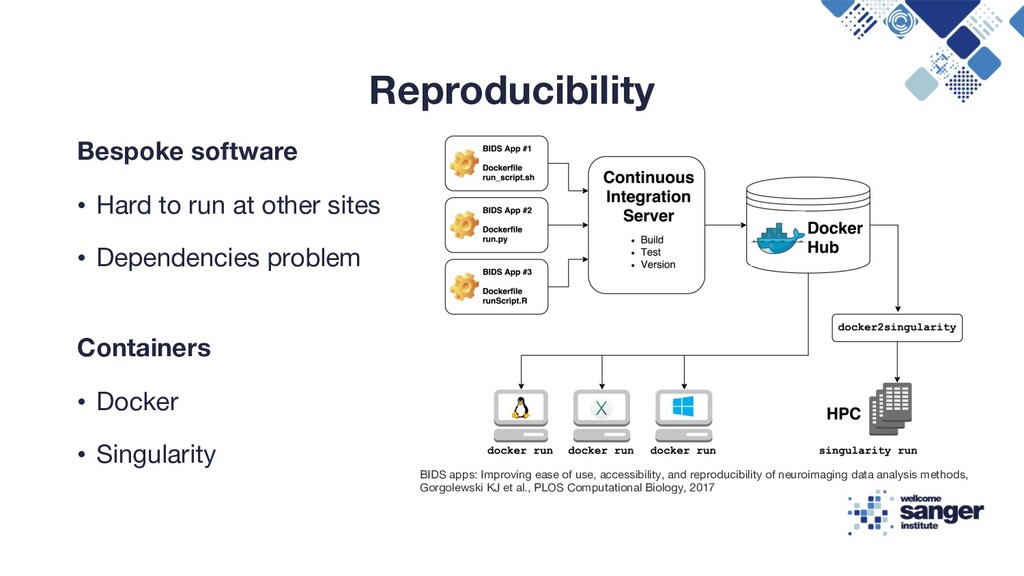
• Dependencies problem Containers • Docker • Singularity BIDS apps: Improving ease of use, accessibility, and reproducibility of neuroimaging data analysis methods, Gorgolewski KJ et al., PLOS Computational Biology, 2017
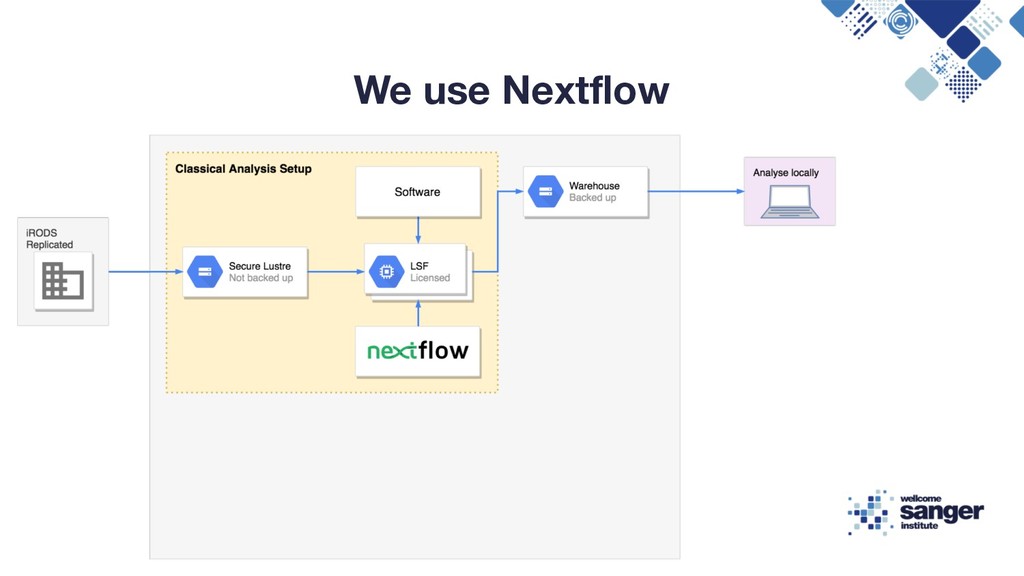
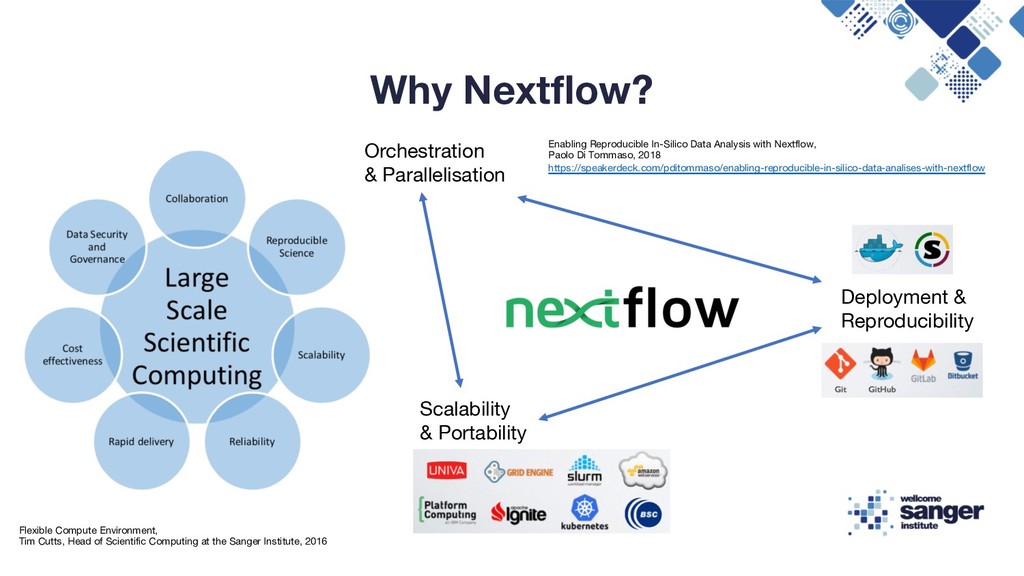
Reproducibility Flexible Compute Environment, Tim Cutts, Head of Scientific Computing at the Sanger Institute, 2016 Enabling Reproducible In-Silico Data Analysis with Nextflow, Paolo Di Tommaso, 2018 https://speakerdeck.com/pditommaso/enabling-reproducible-in-silico-data-analises-with-nextflow
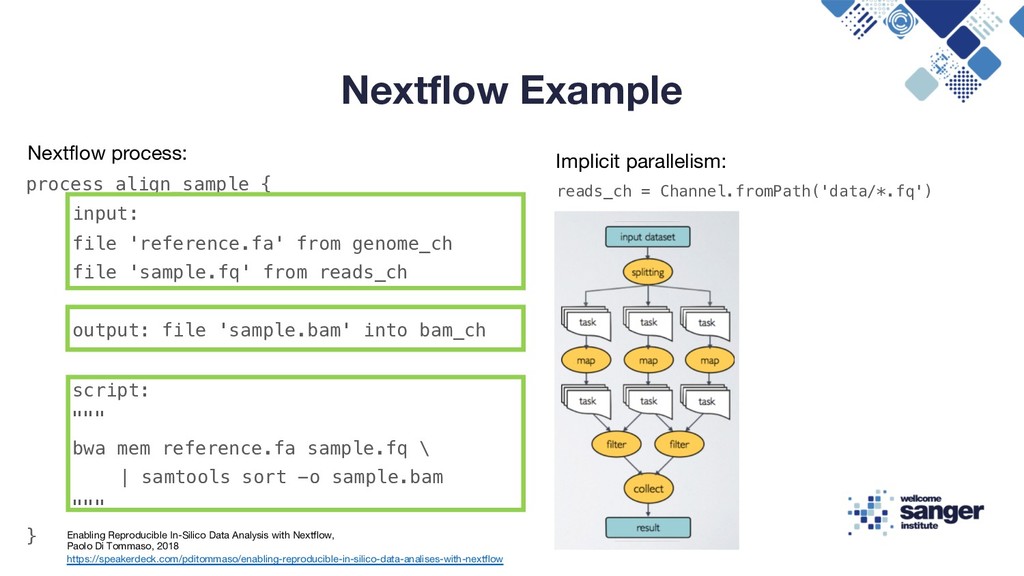
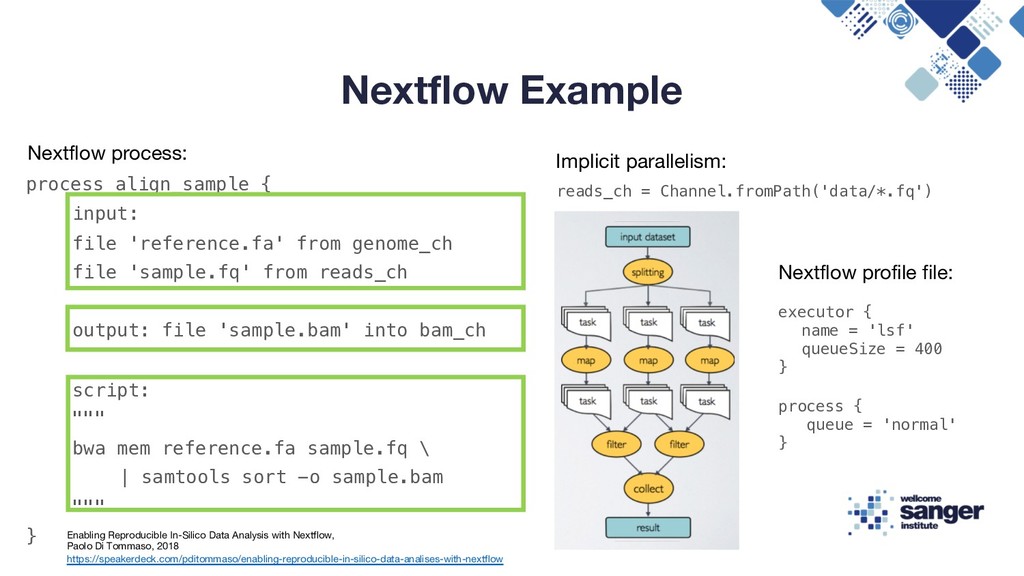
-o sample.bam Enabling Reproducible In-Silico Data Analysis with Nextflow, Paolo Di Tommaso, 2018 https://speakerdeck.com/pditommaso/enabling-reproducible-in-silico-data-analises-with-nextflow
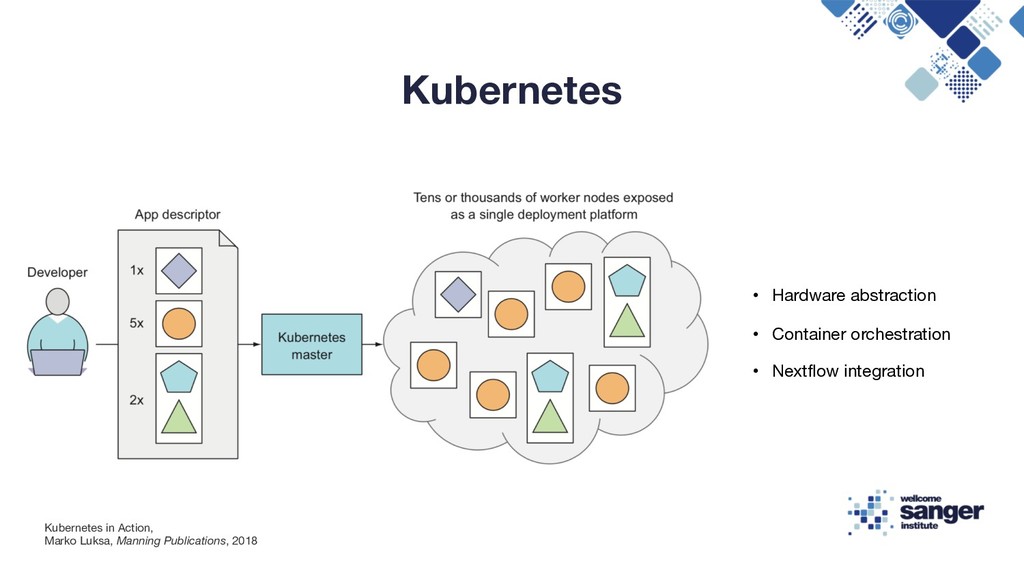
a better way of deploying and managing their software/infrastructure to scale globally • In ~2004 Google developed Borg and then Omega and kept it secret until 2014 • In 2014 Google introduced Kubernetes, an open-source system based on the experience gained through Borg, Omega • Kubernetes facilitates the deployment and serving of web applications
• Provides containerized R/python/Julia environments • Rstudio server is included • Big compute in a browser window • We create templates for downstream analysis https://github.com/cellgeni/notebooks • Installation on Kubernetes cluster (one liner): helm upgrade --install jpt jupyterhub/jupyterhub --namespace jpt --version 0.7.0-beta.2 --values jupyter-config.yaml
Downstream analysis pipelines • Ready to be used • User-friendly • Installation on Kubernetes cluster is also a one liner https://github.com/ebi-gene-expression-group/container-galaxy-sc-tertiary
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
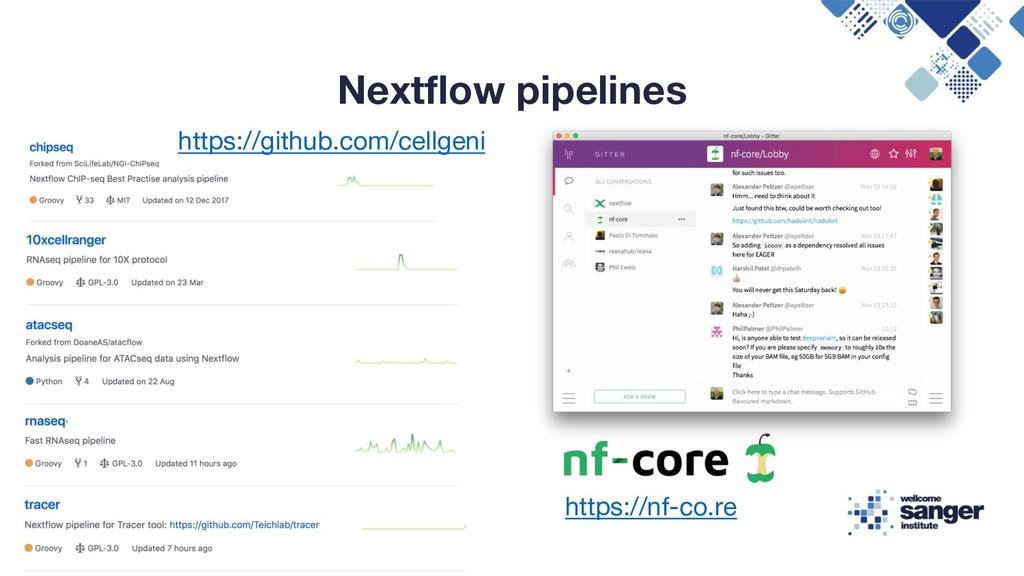{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
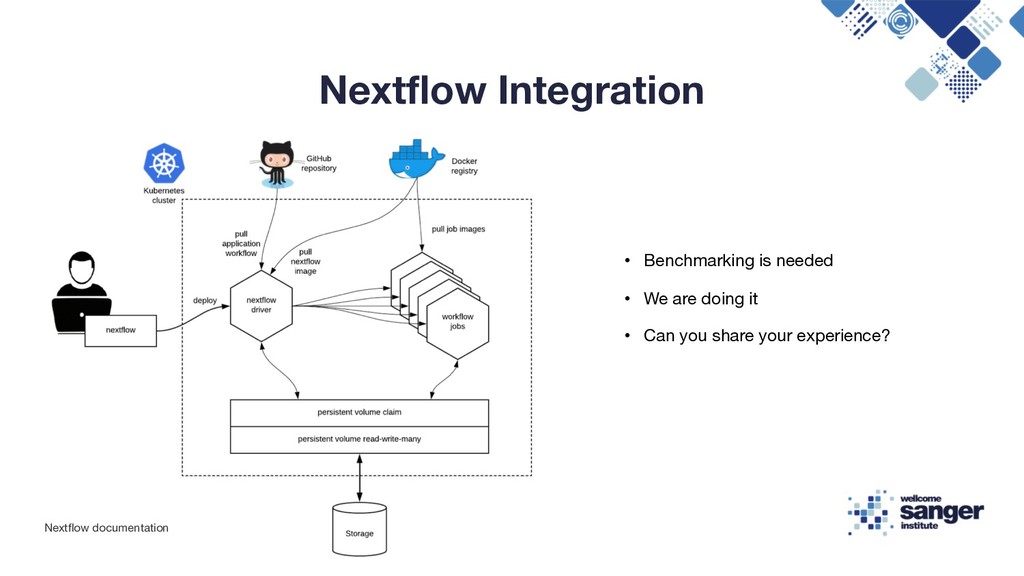
{kind=link}
{kind=link}
{kind=link}
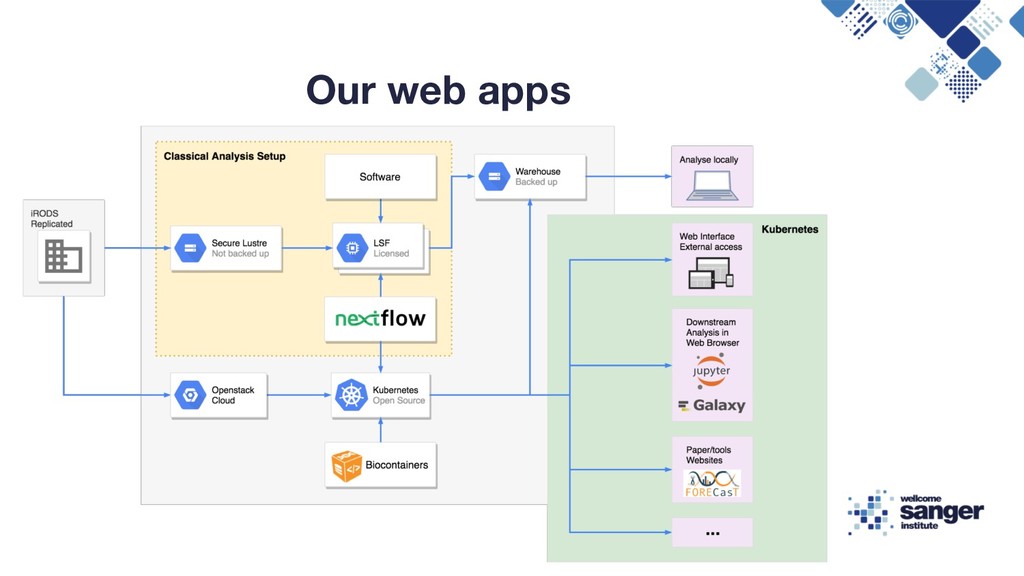{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}