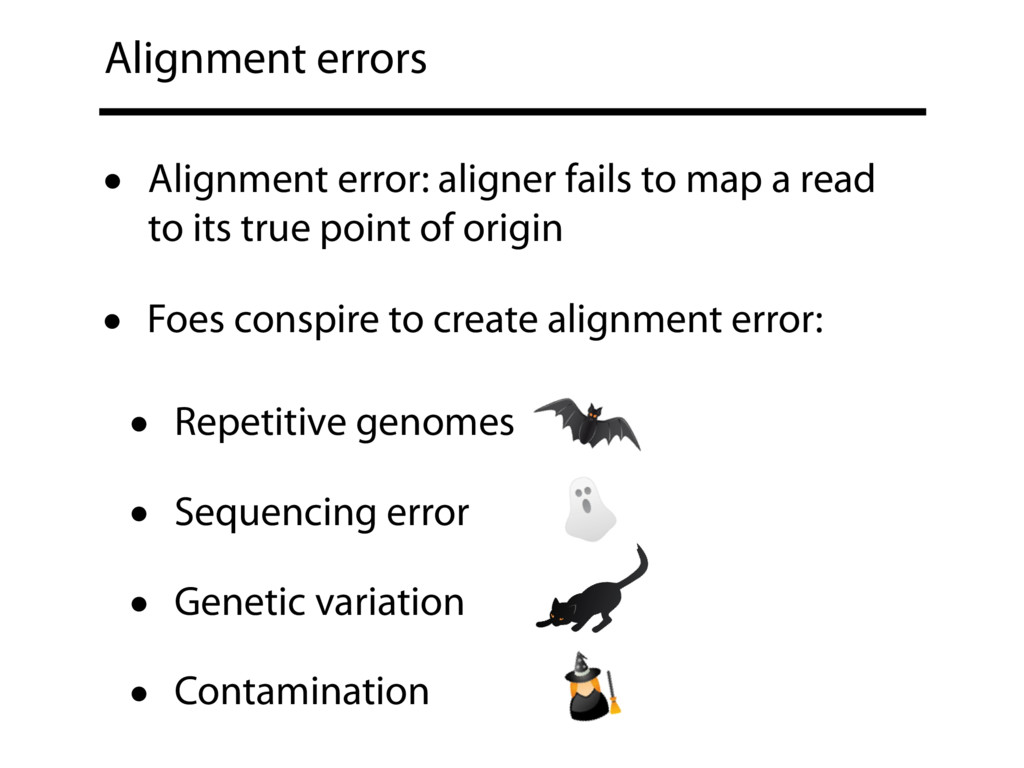
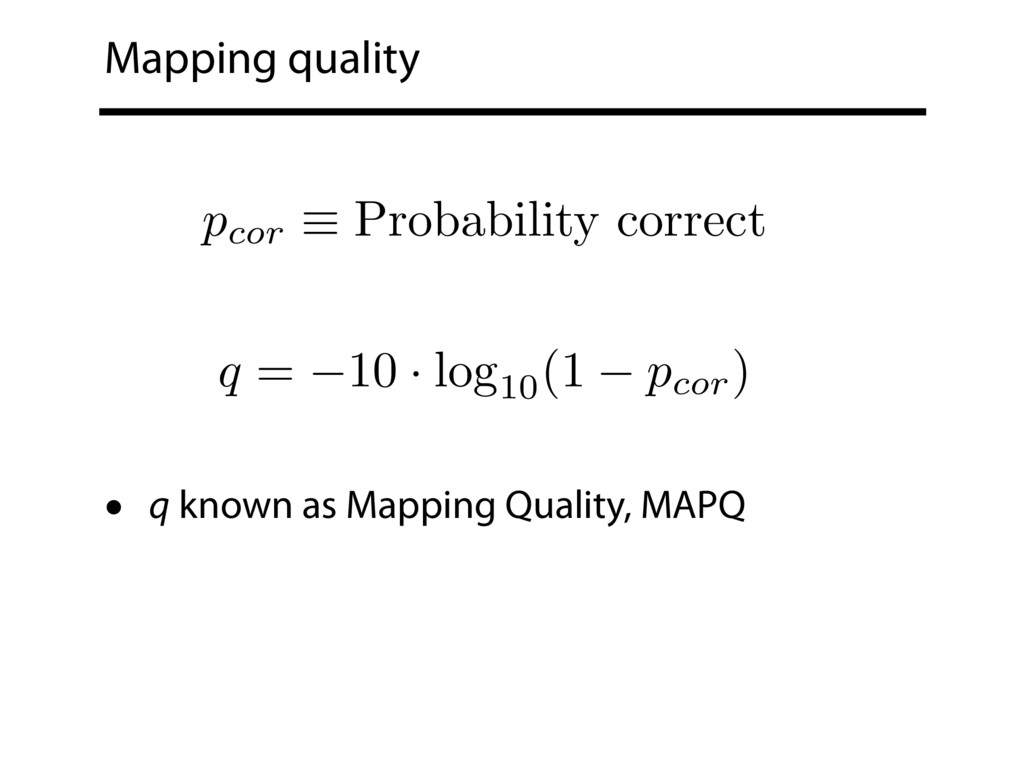
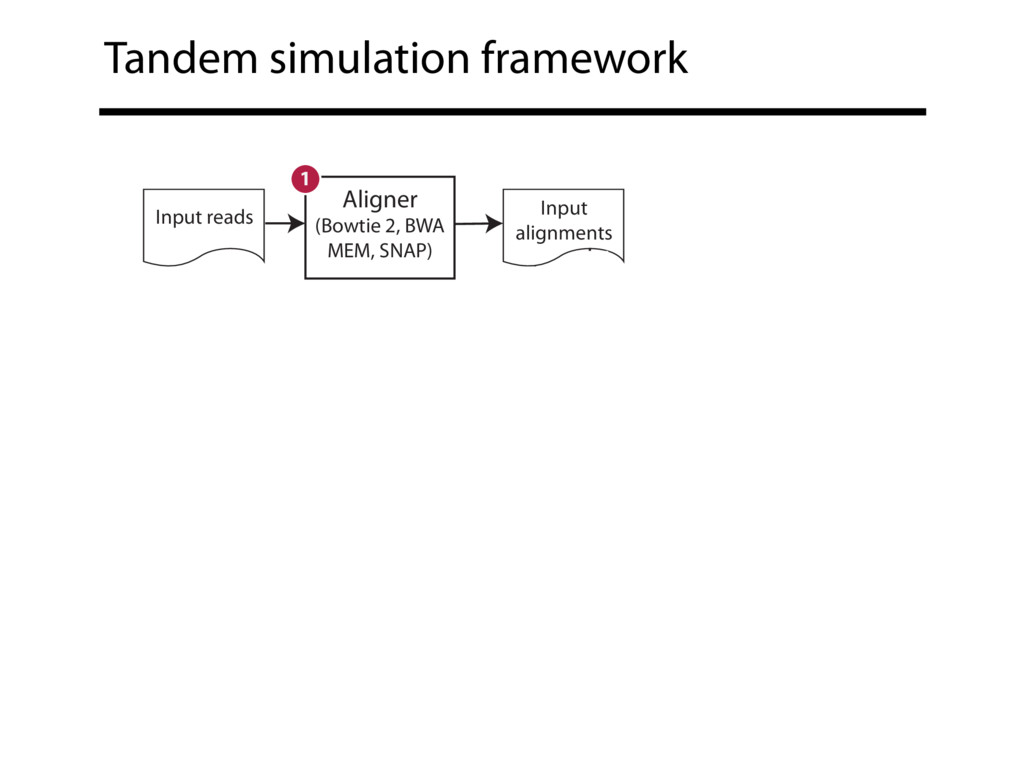
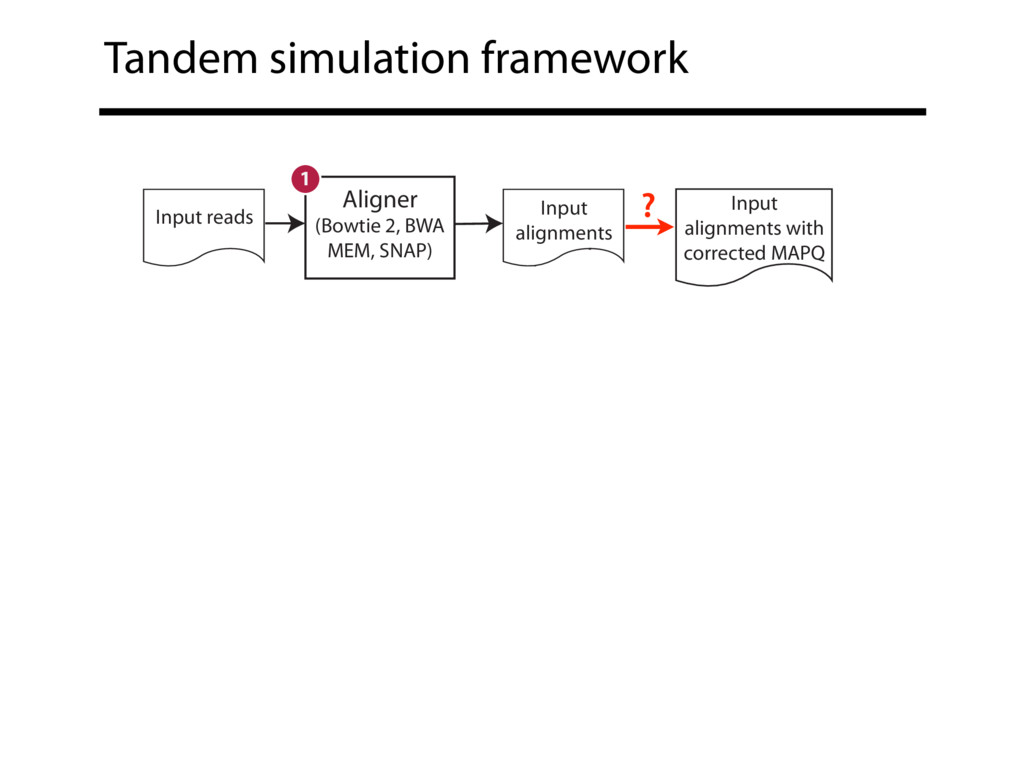
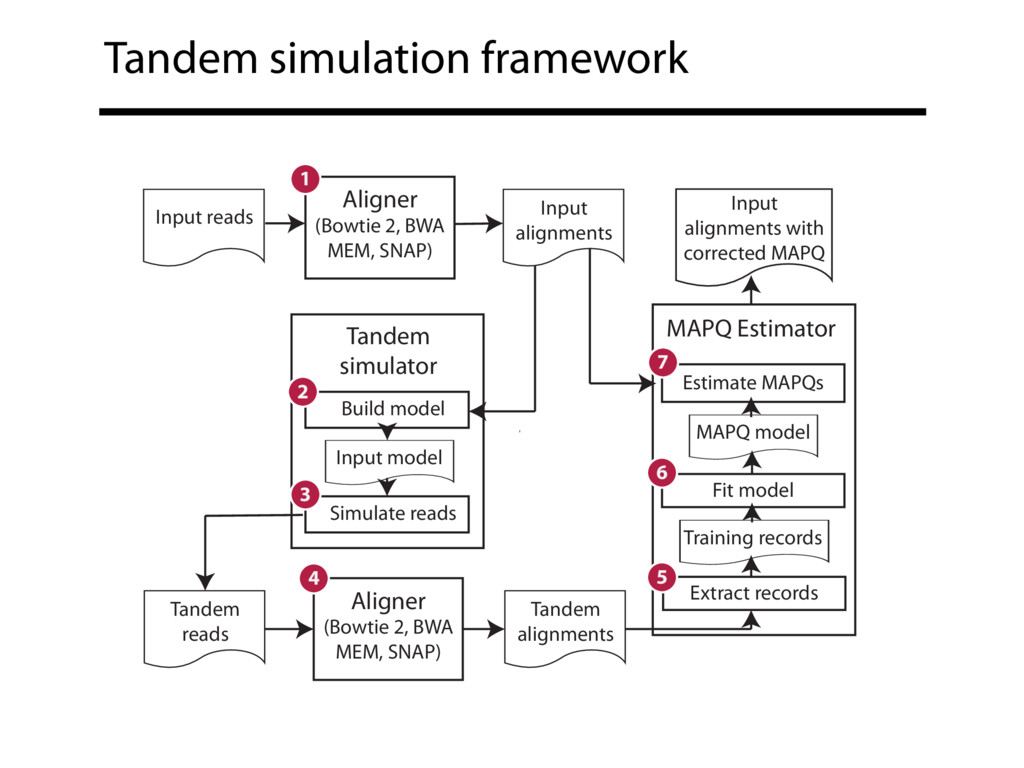
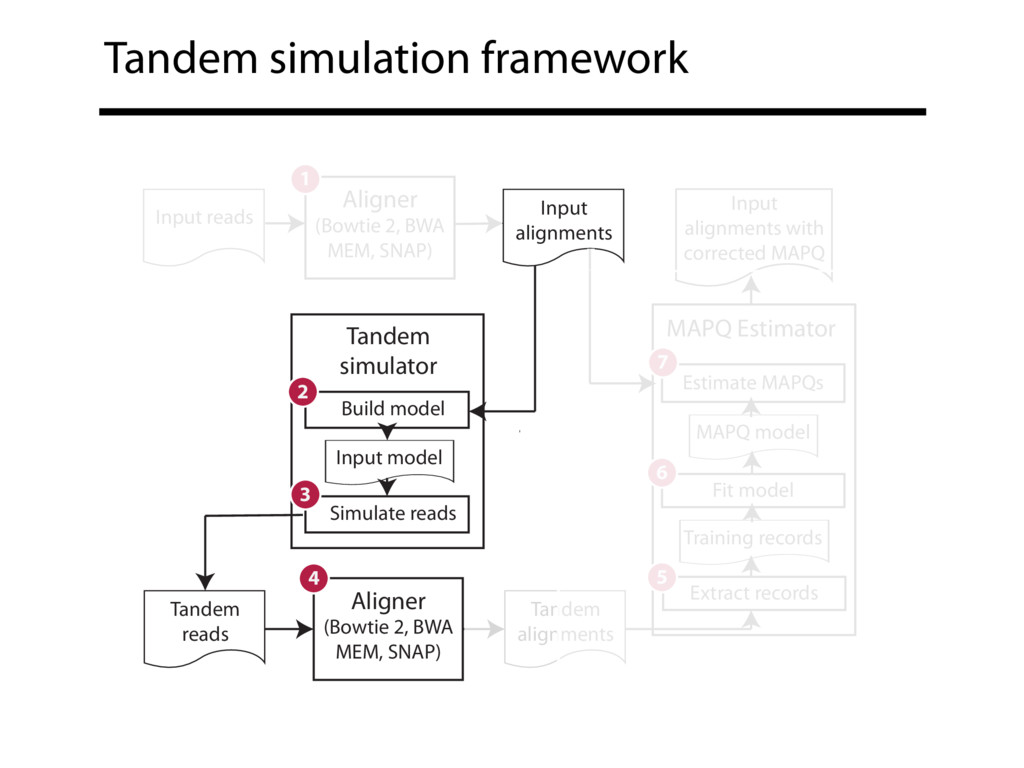
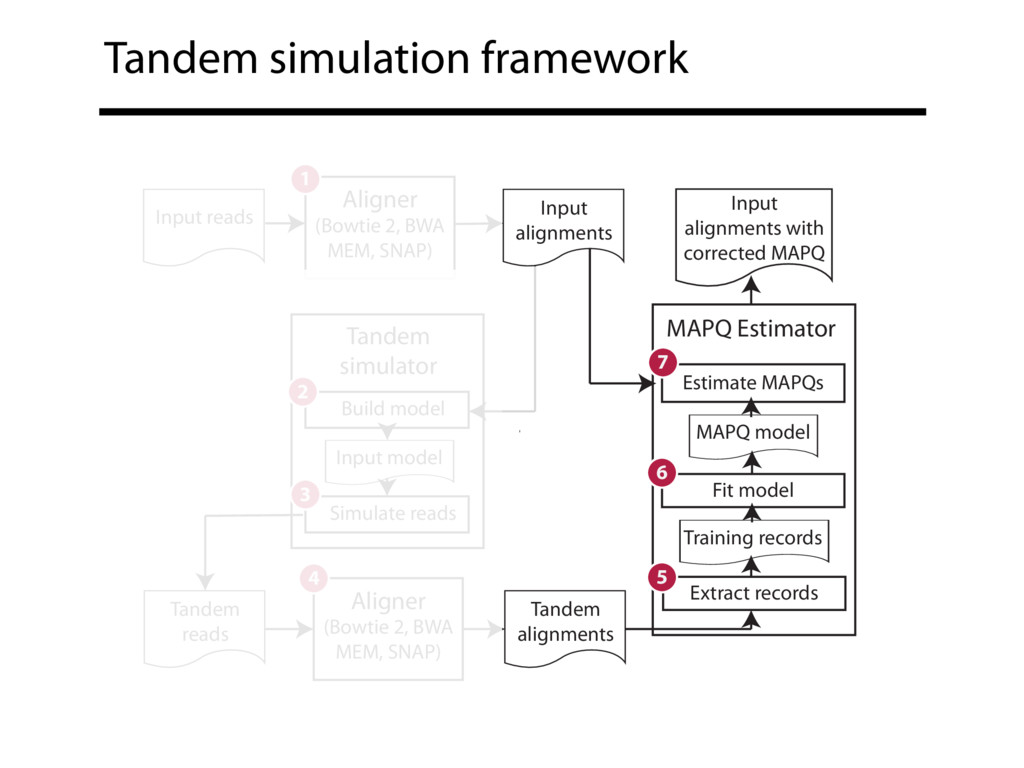
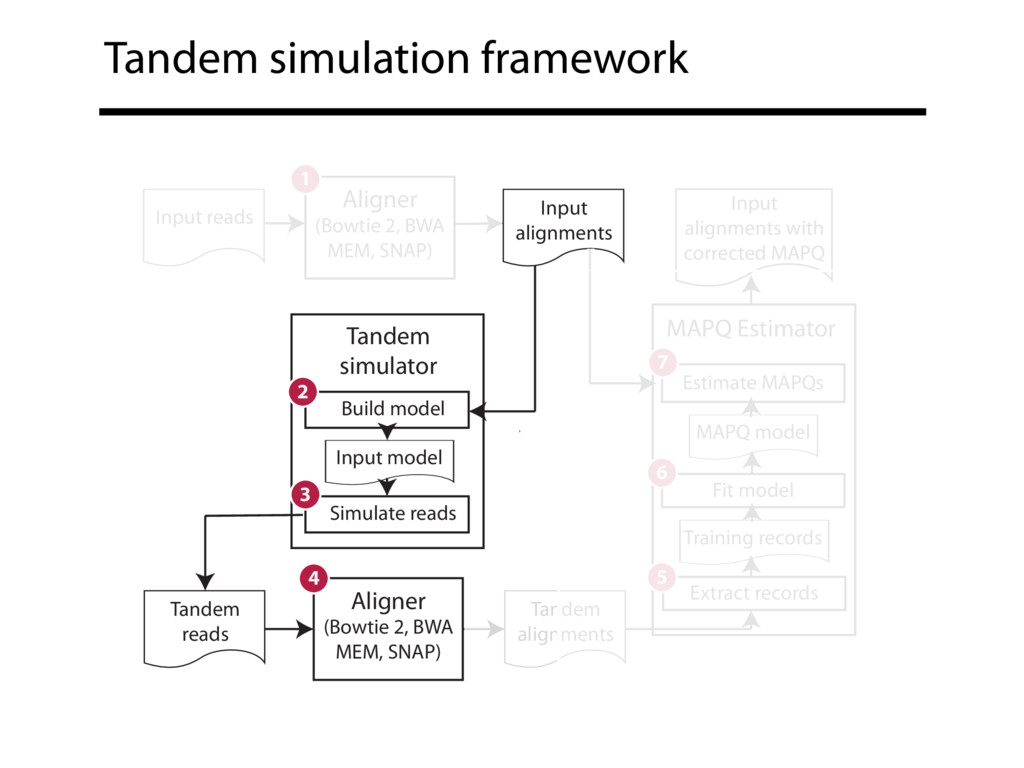
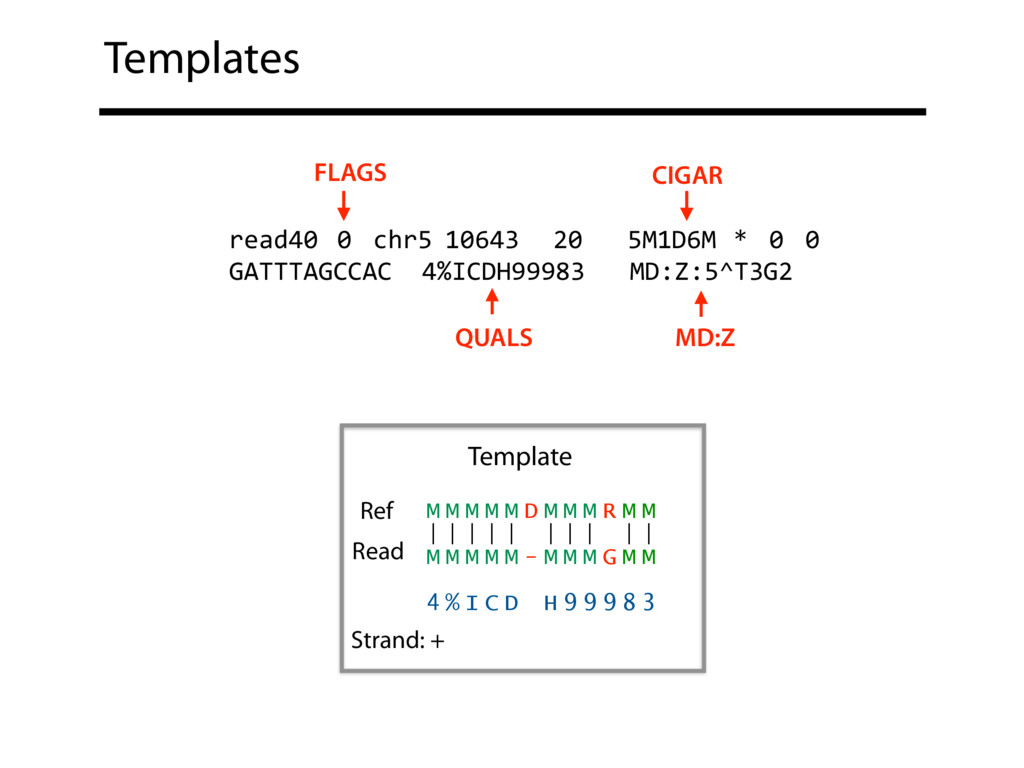
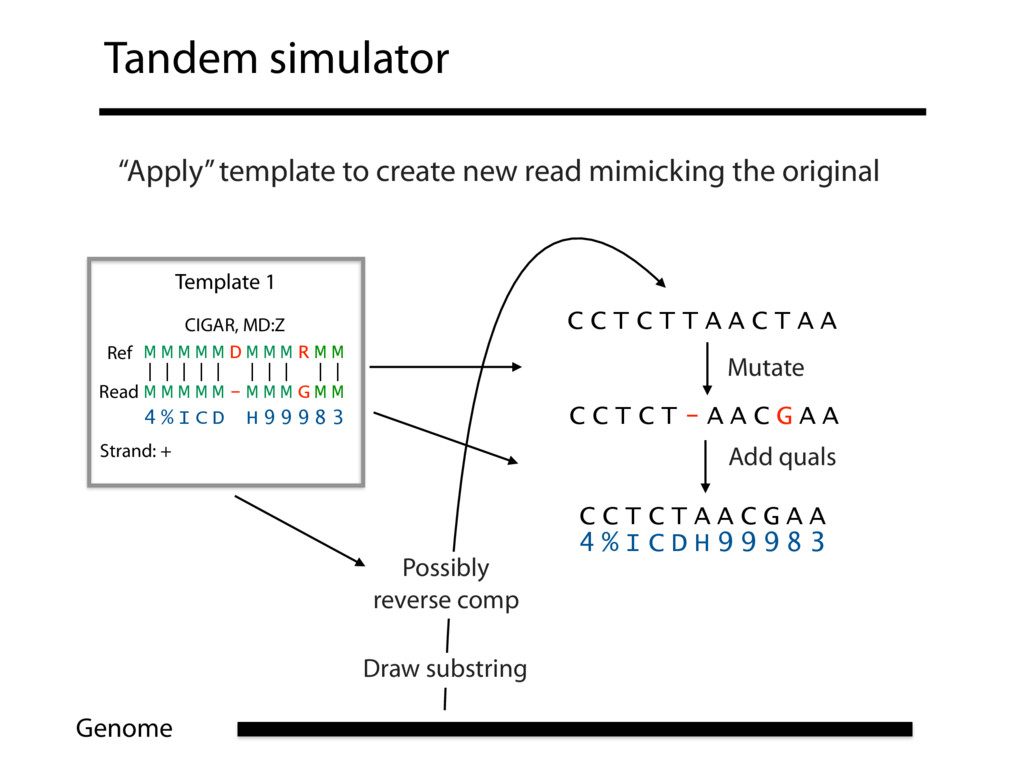
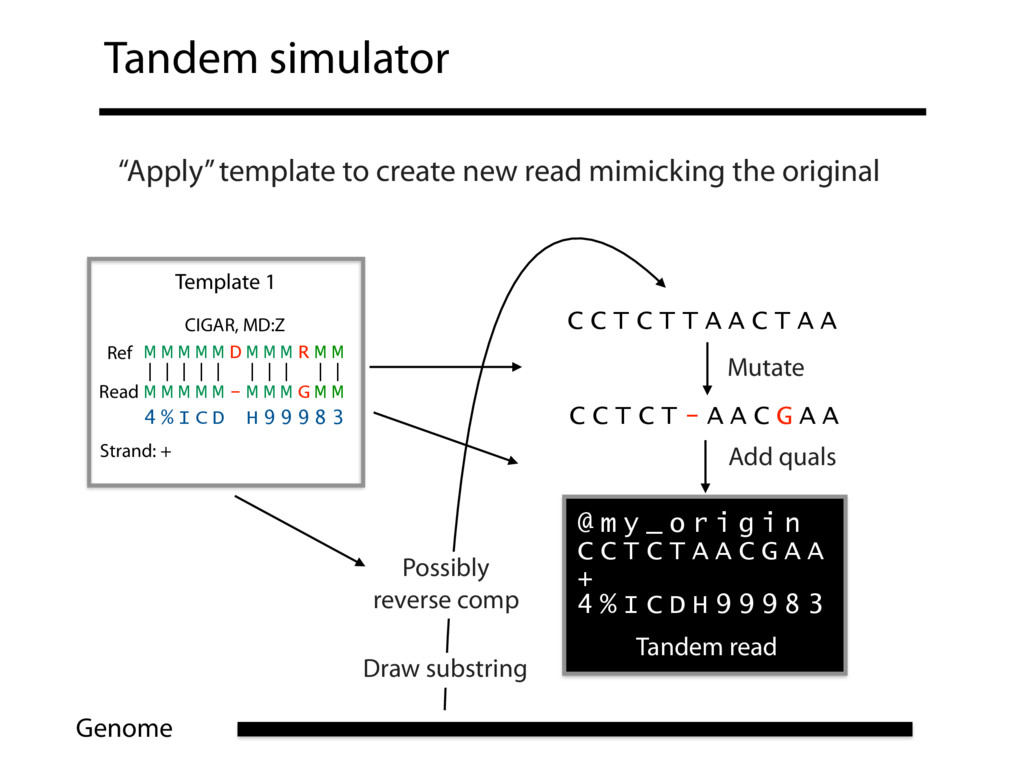
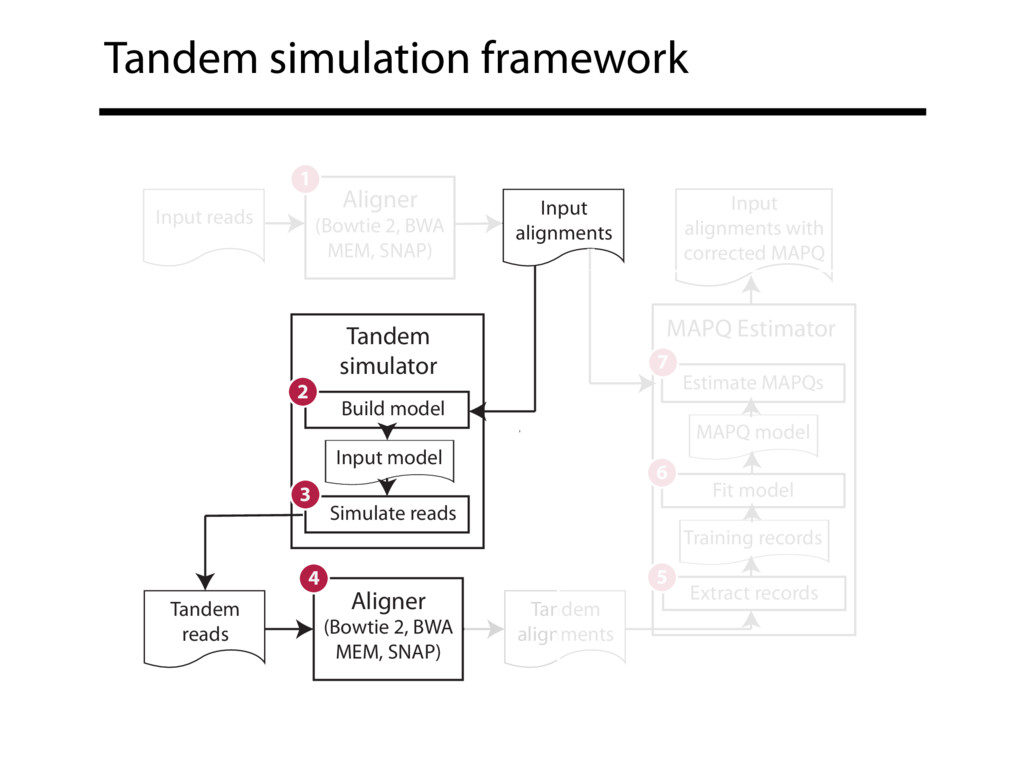
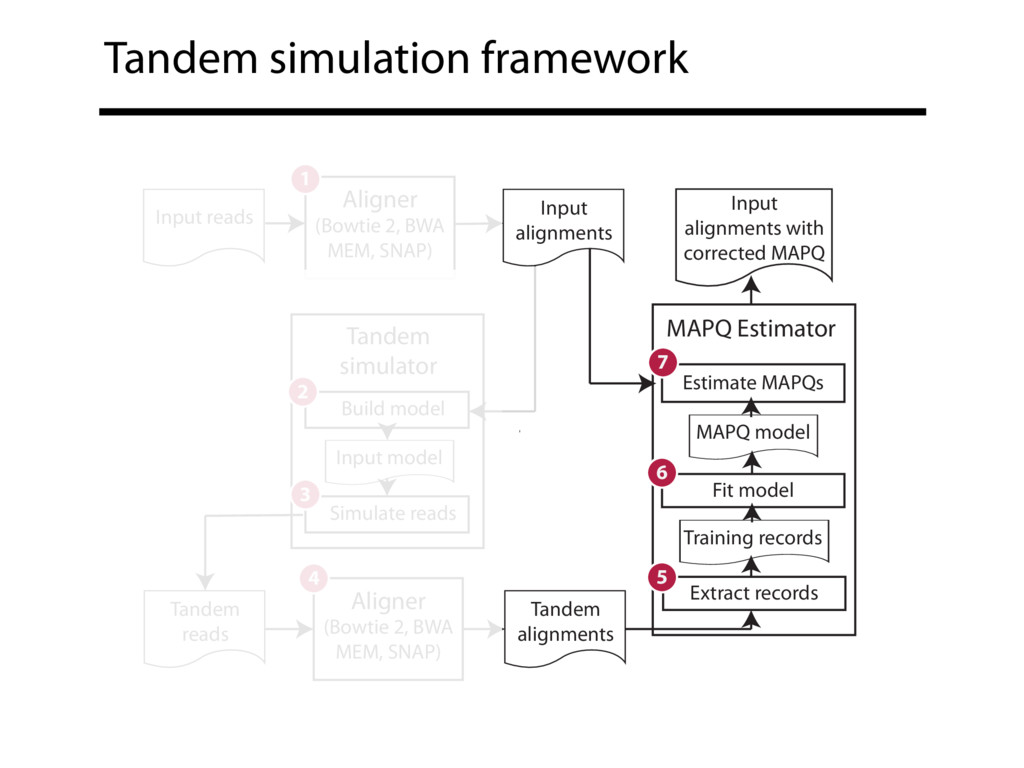
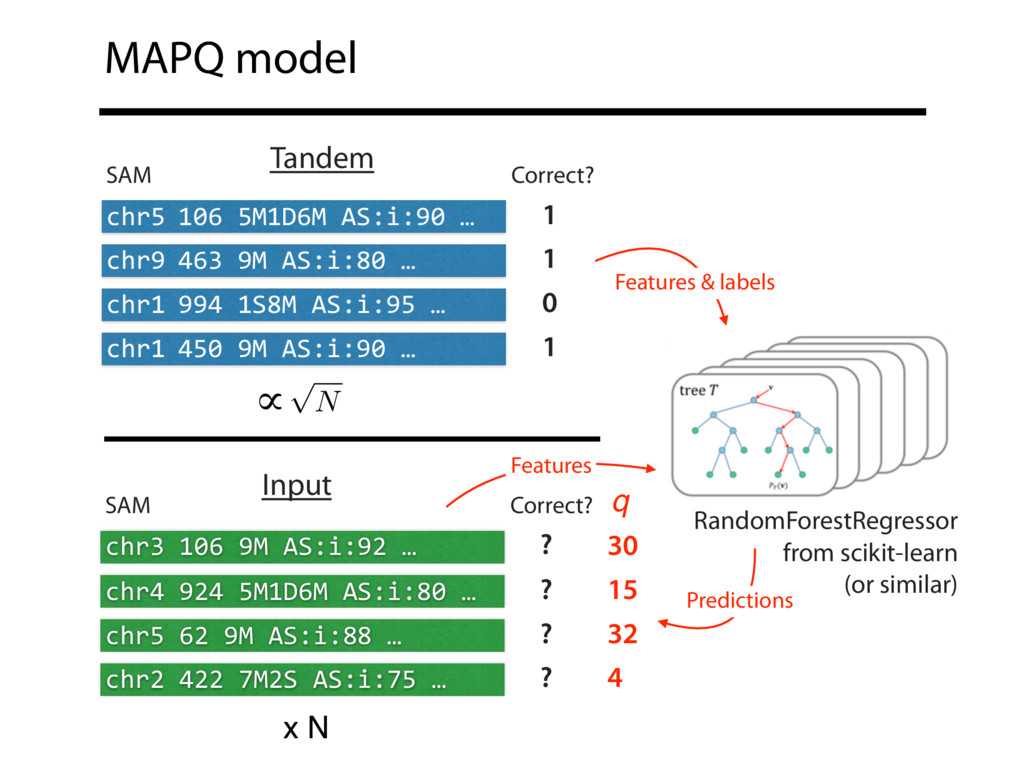
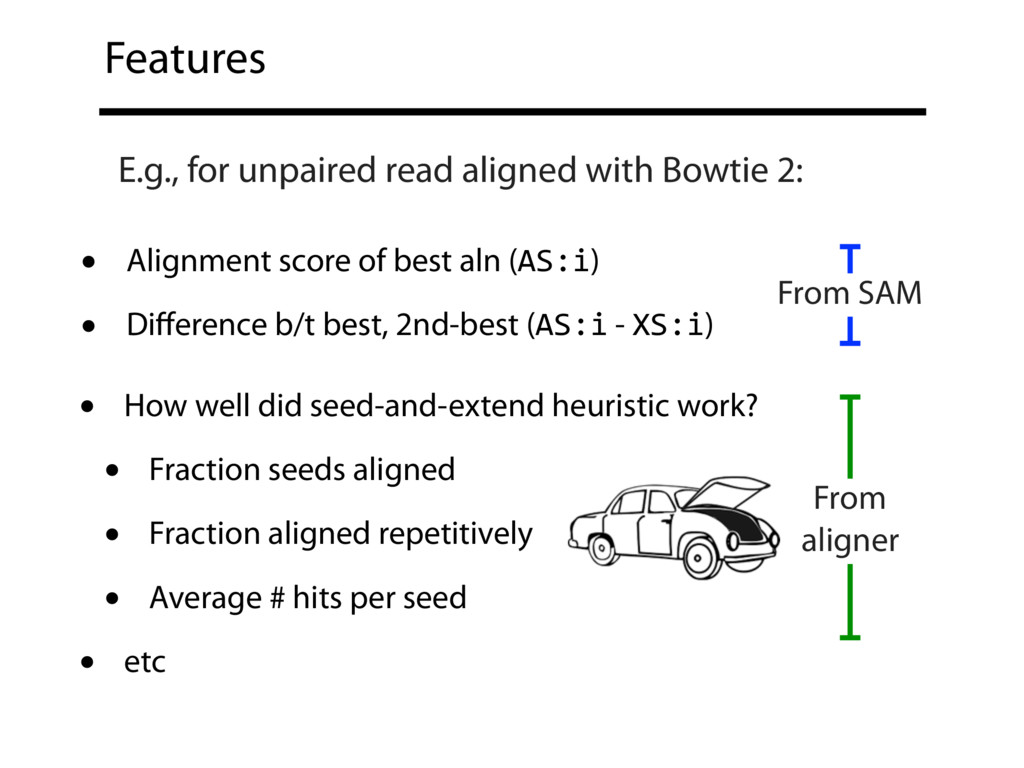
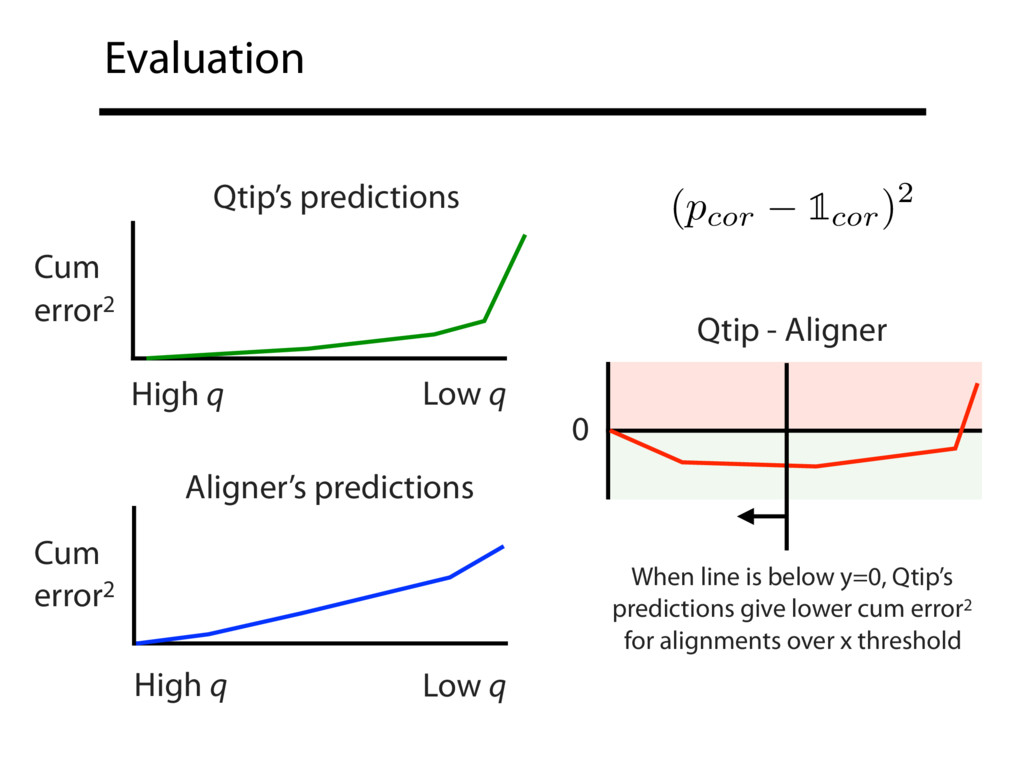
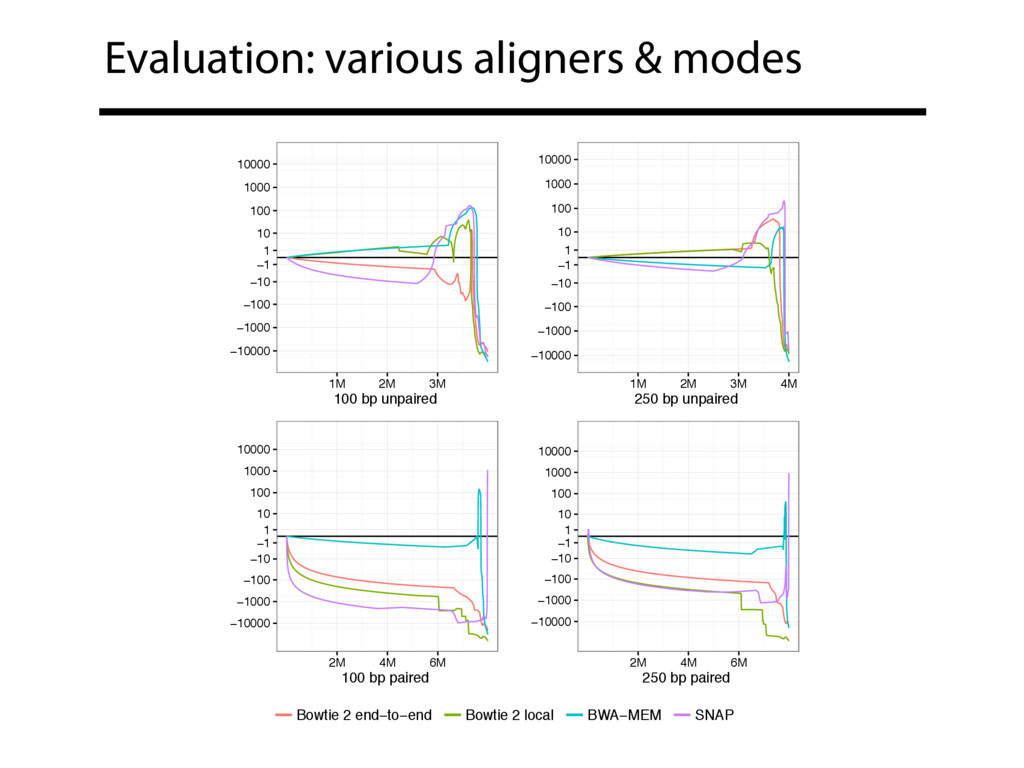
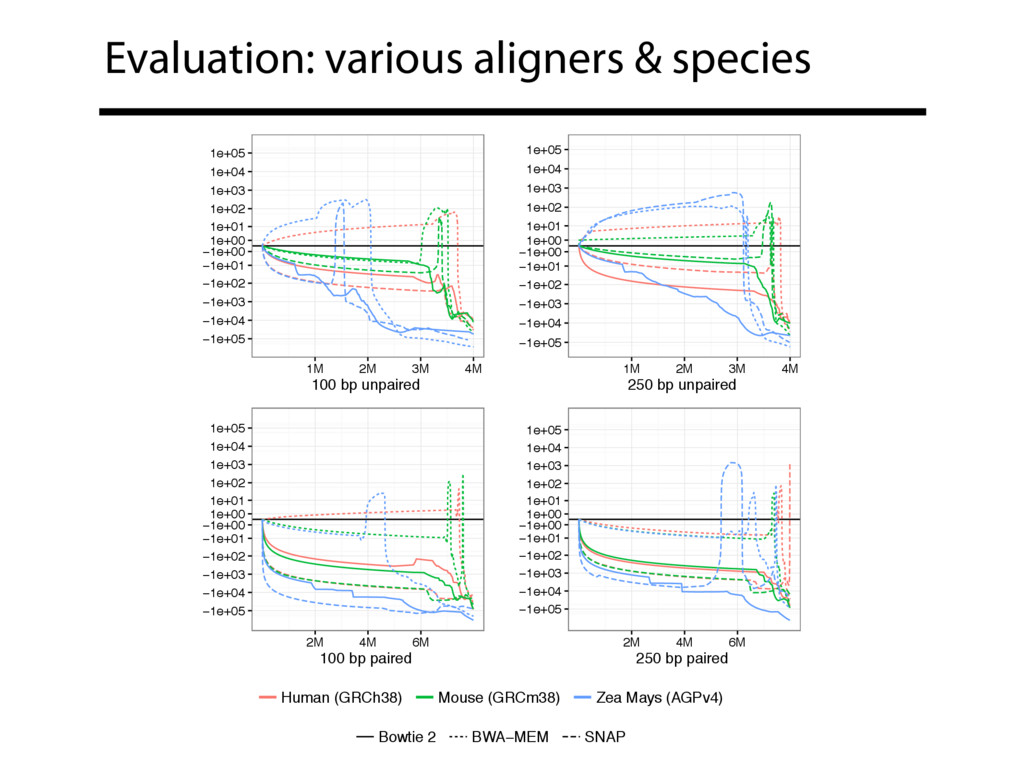
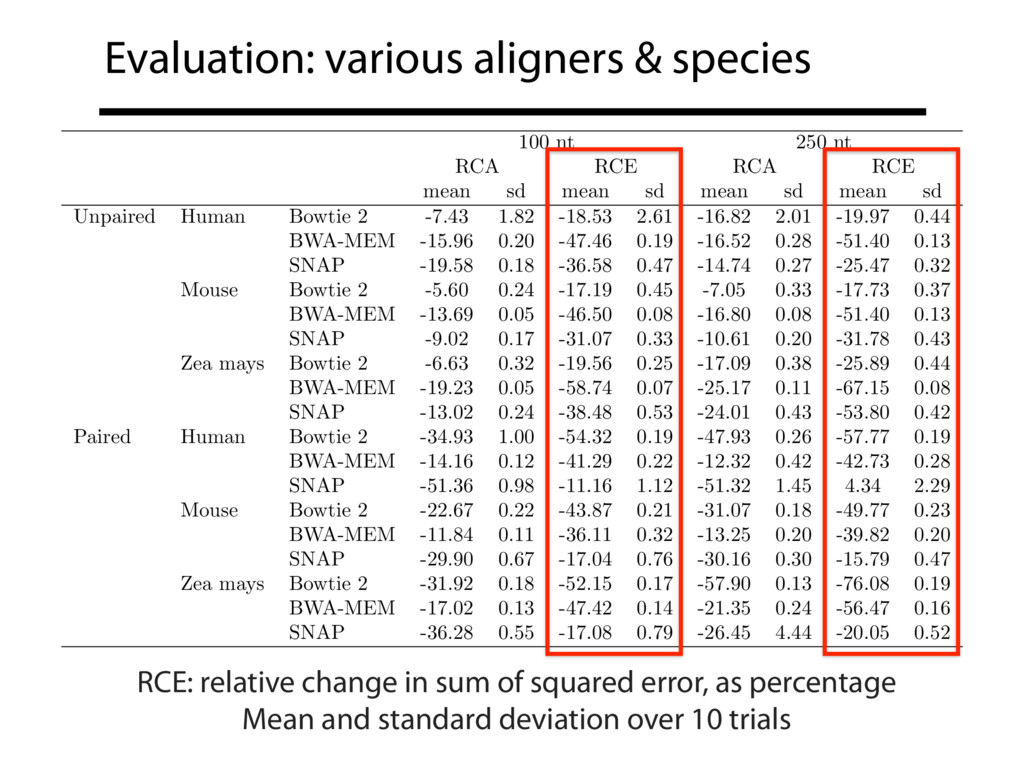
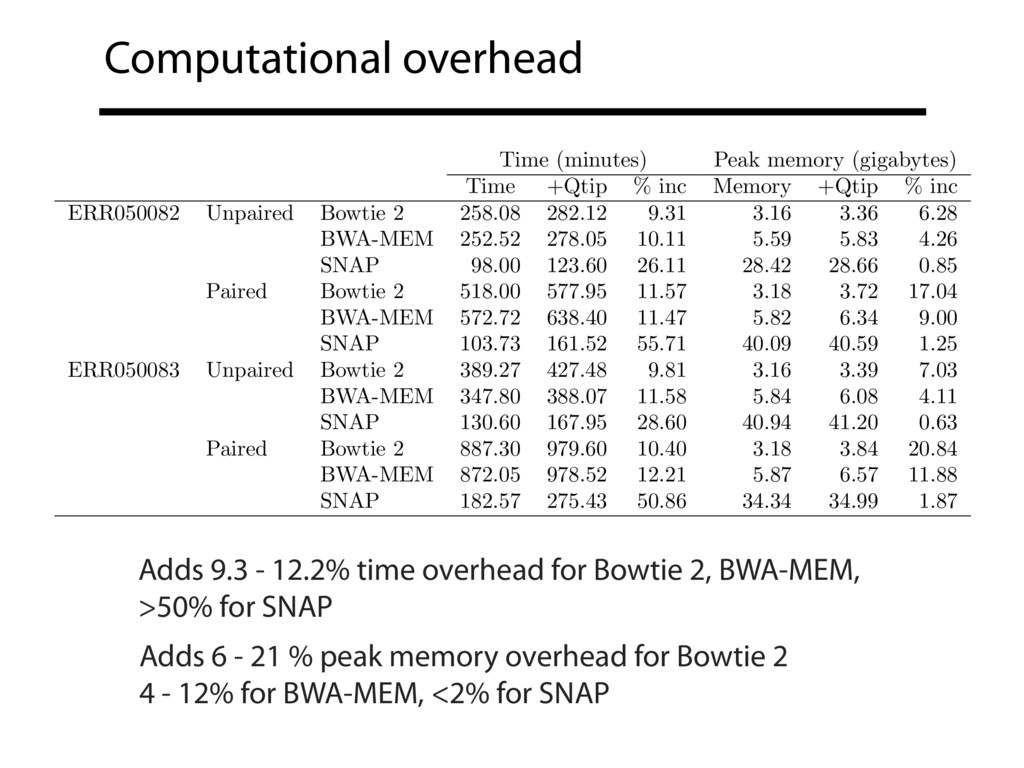
Read alignment is the first step in most sequencing data analyses. It is also a source of errors and interpretability problems. Repetitive genomes, algorithmic shortcuts, and genetic variation impede the aligner's ability to find a read's true point of origin. Aligners therefore report a mapping quality: the probability the reported point of origin for a read is incorrect. However, there are no proposals for how to calculate mapping qualities in a general way, applicable across tools and alignment scenarios. I describe an accurate, aligner-agnostic framework for predicting mapping qualities that works by simulating a set of "tandem" reads, similar to the input reads in important ways, but for which the true point of origin is known. Alignments of tandem reads are used to build a model for predicting mapping quality, which is then applied to the input-read alignments. The model is automatically tailored to the alignment scenario at hand, allowing it to make accurate mapping-quality predictions across a range of read lengths, alignment parameters, genomes, and read aligners. I implement this approach in a software tool called Qtip, which is accurate, low-overhead, and compatible with popular read aligners. Qtip is open source software available from https://github.com/BenLangmead/qtip and experiments are published at https://github.com/BenLangmead/qtip-experiments.
![Ben Langmead Assistant Professor, JHU Computer Science [email protected], www.langmead-lab.org, @BenLangmead](https://files.speakerdeck.com/presentations/7cf9999490c348498da3955314a9566d/slide_0.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}