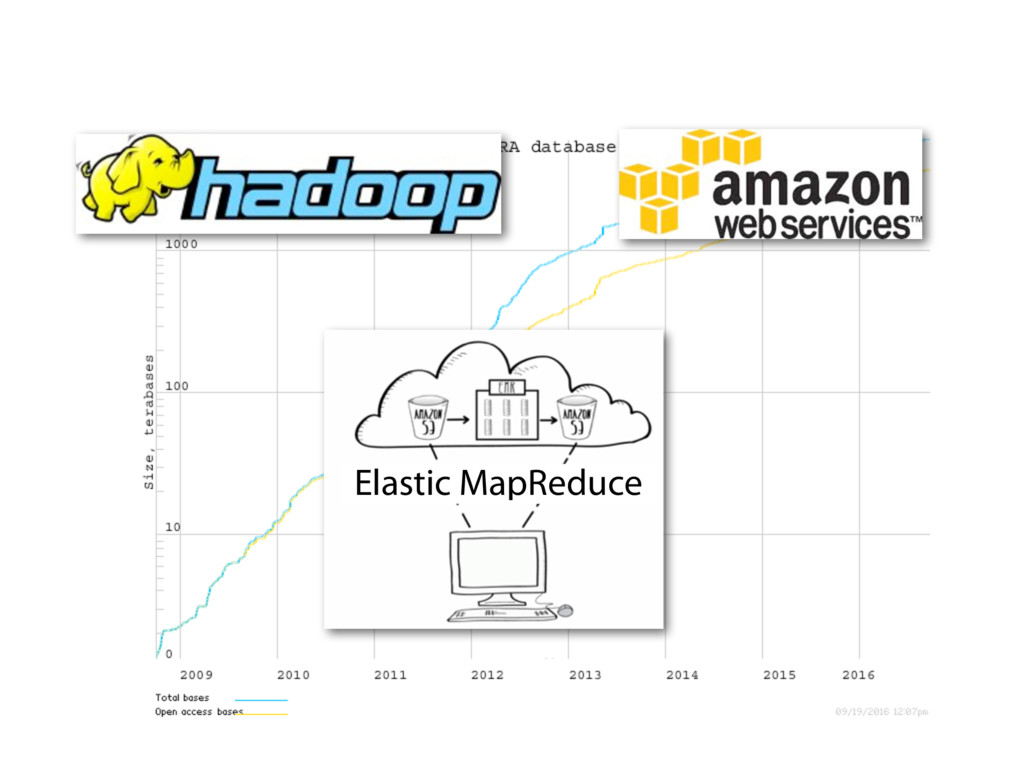
RNA sequencing is a ubiquitous tool for assaying gene expression. Public sequencing data archives such as the Sequence Read Archive now hold more than 50,000 human RNA-seq samples, and the size of the archive doubles approximately every 18 months. Many of these archived studies are valuable to biological researchers and methods developers. However, samples are available only as compressed collections of raw data. Processing the raw data into a form suitable for various downstream analyses is challenging. Care is required to craft summaries that are both concise — convenient for researchers to download and interact with — and useful in a variety of downstream scenarios.
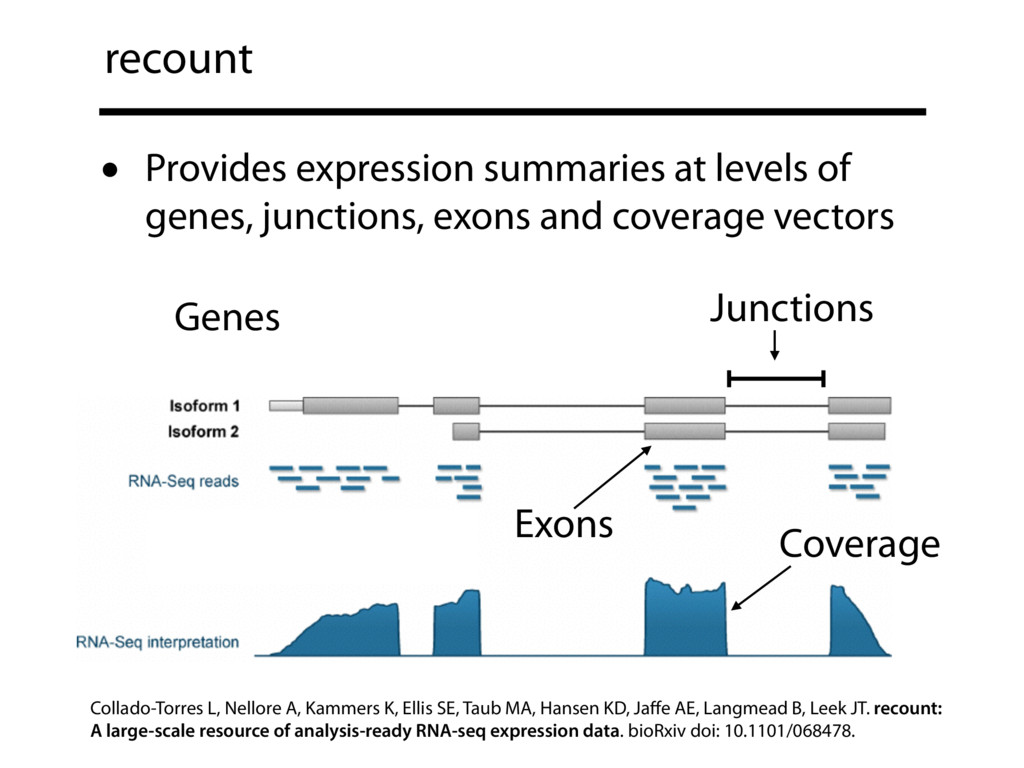
The recount resource addresses this issue by summarizing a huge amount of public data — about 50,000 human RNA-seq samples from the Sequence Read Archive (SRA) and almost 10,000 samples from the GTEx project — into a form that is easy to query. recount is hosted on SciServer, and takes advantage of R and Jupyter notebooks to make it easy for anyone to query the summarized data. Here we exhibit these data summaries — compiled at the level of genes, exons, exon-exon-junctions and base-level coverage — as well as how to use the SciServer Jupyter notebook interface to perform sophisticated analyses.
![Ben Langmead Assistant Professor, Computer Science [email protected] IDIES Symposium, October](https://files.speakerdeck.com/presentations/6186fd0ca2a04d499147cfd45000ec47/slide_0.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}