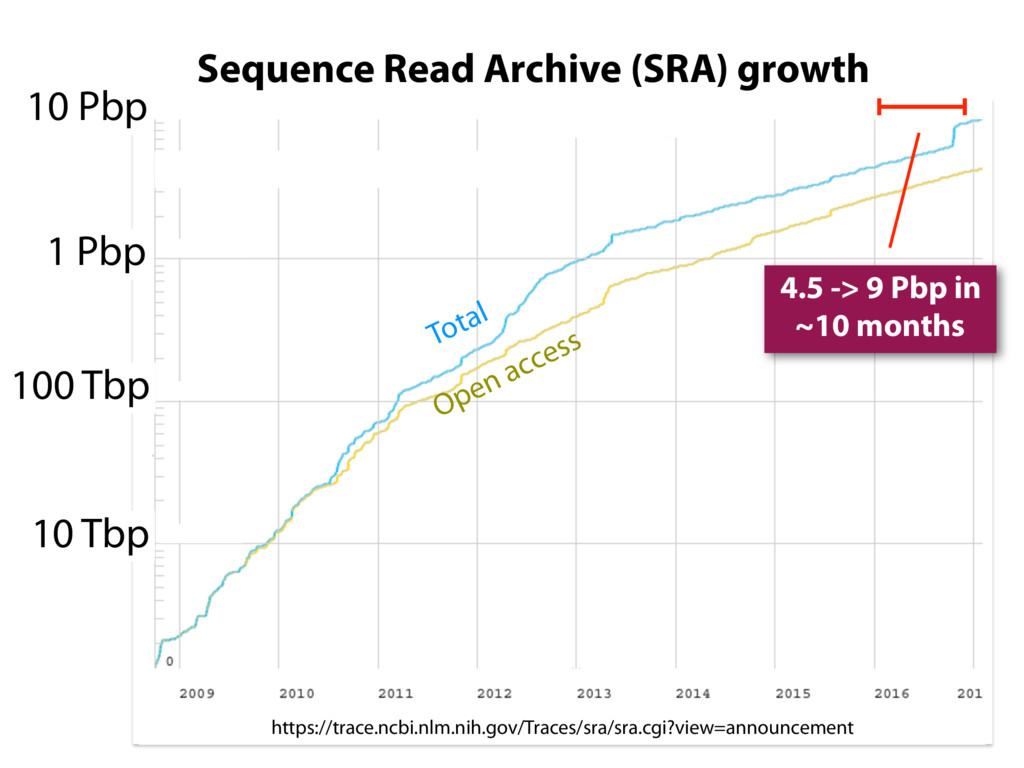
Abstract: The Sequence Read Archive contains RNA-seq data for over 450K samples, including over 140K from humans. Large-scale projects like GTEx and ICGC are generating RNA-seq data on many thousands of samples. Such huge datasets are valuable, but unwieldy for typical researchers. I will describe work toward the goal of making it easy for researchers to use the archived RNA-seq data available today. I will highlight Rail-RNA (http://rail.bio), its dbGaP-protected version (http://docs.rail.bio/dbgap/), as well as the recount resource (https://jhubiostatistics.shinyapps.io/recount/) and Snaptron service/API (http://snaptron.cs.jhu.edu). Besides showcasing these tools and resources, I'll expound three themes: (a) pulic data is valuable but not easy to use and computationalists should attack this; (b) scalability is not just about scaling software to be distributed & multi-threaded, but is also about making the best use of many datasets at once; (c) "strategically unplugging" from gene annotations can lead to clearer statements about splicing and differential expression.
This is joint work with Abhinav Nellore, Jeff Leek, Kasper Hansen, Andrew Jaffe and others.
![Ben Langmead Assistant Professor, JHU Computer Science [email protected], langmead-lab.org, @BenLangmead](https://files.speakerdeck.com/presentations/626d44722d114a34bf874c509d3b4594/slide_0.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
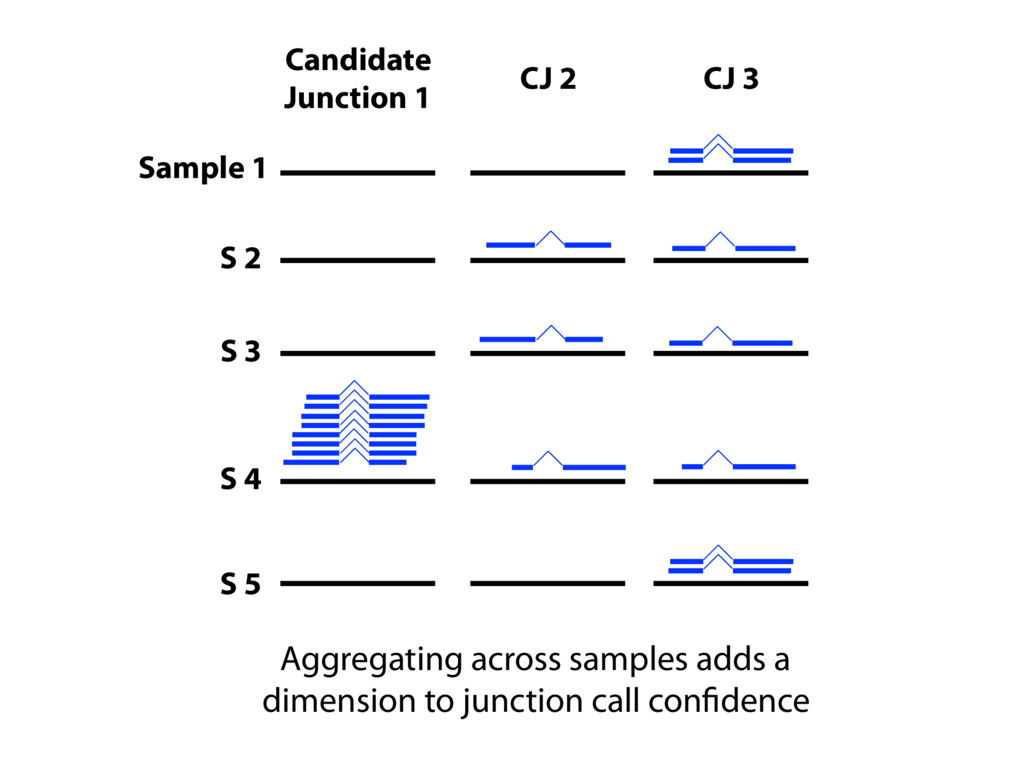{kind=link}
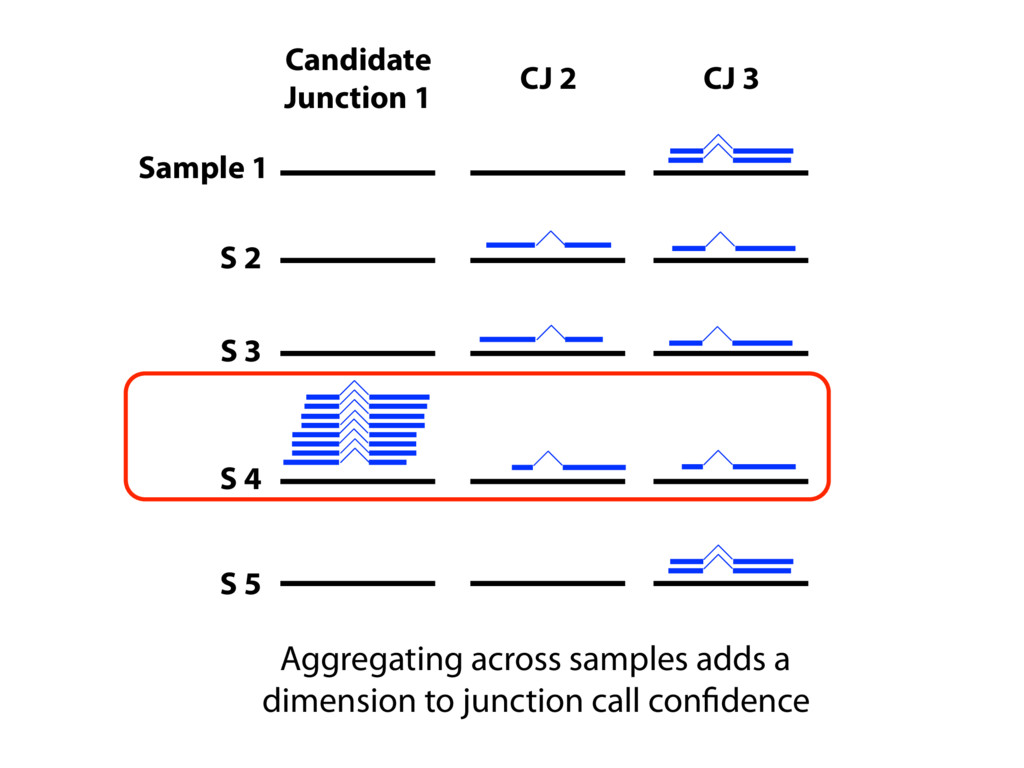{kind=link}
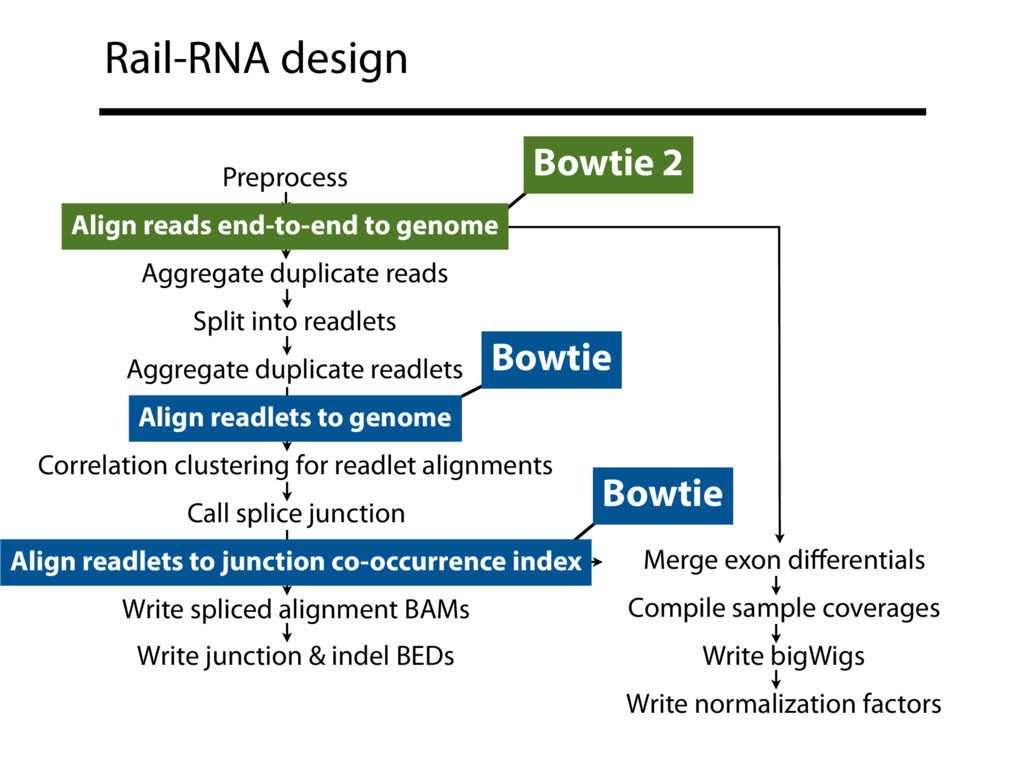{kind=link}
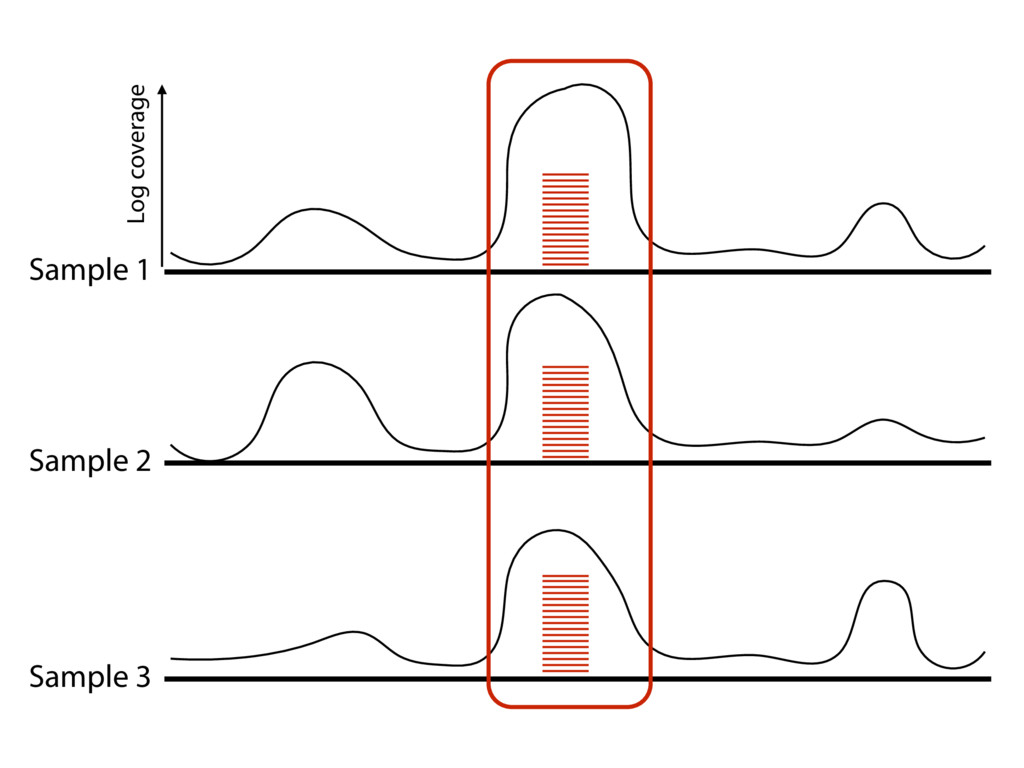{kind=link}
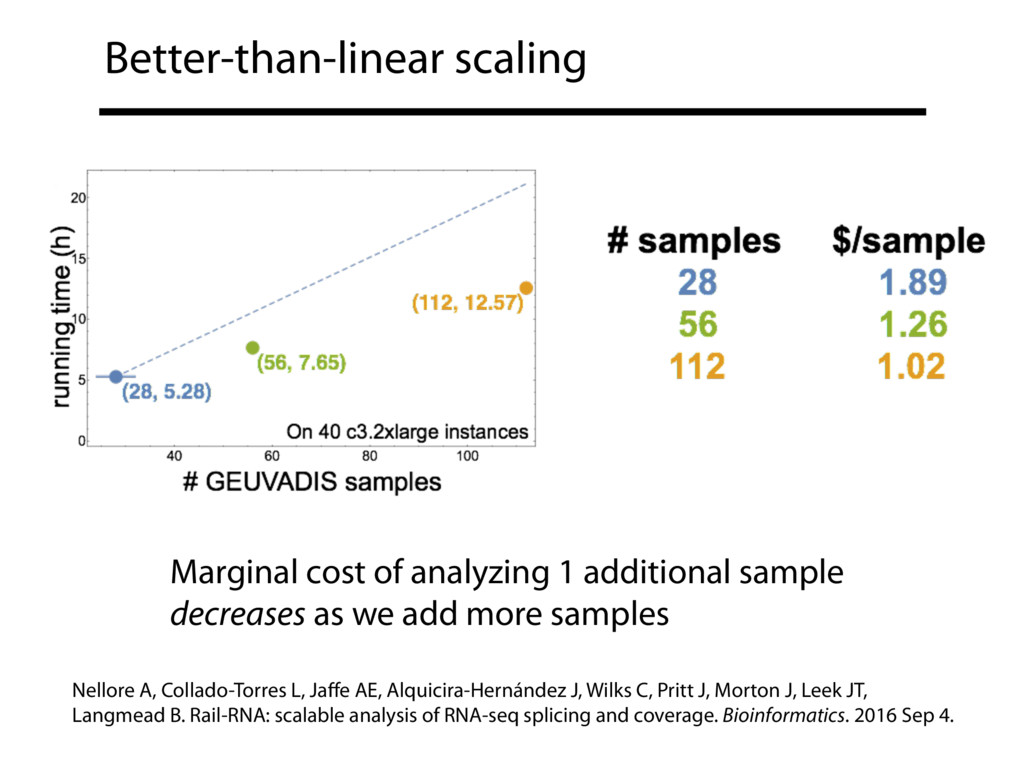{kind=link}
{kind=link}
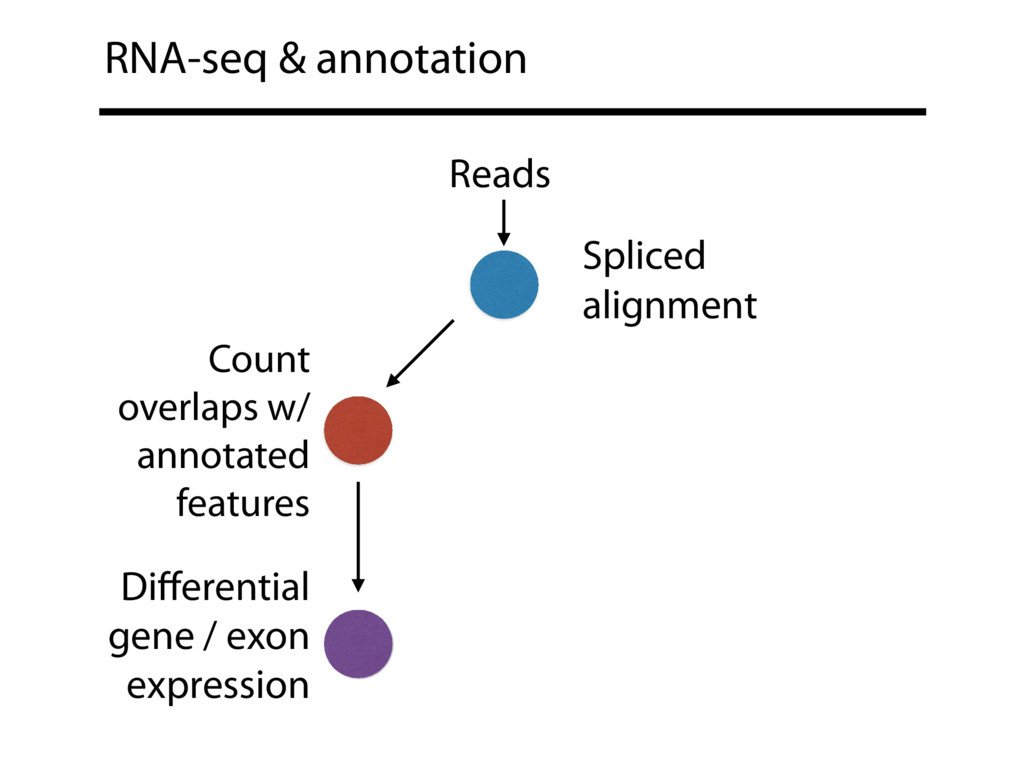{kind=link}
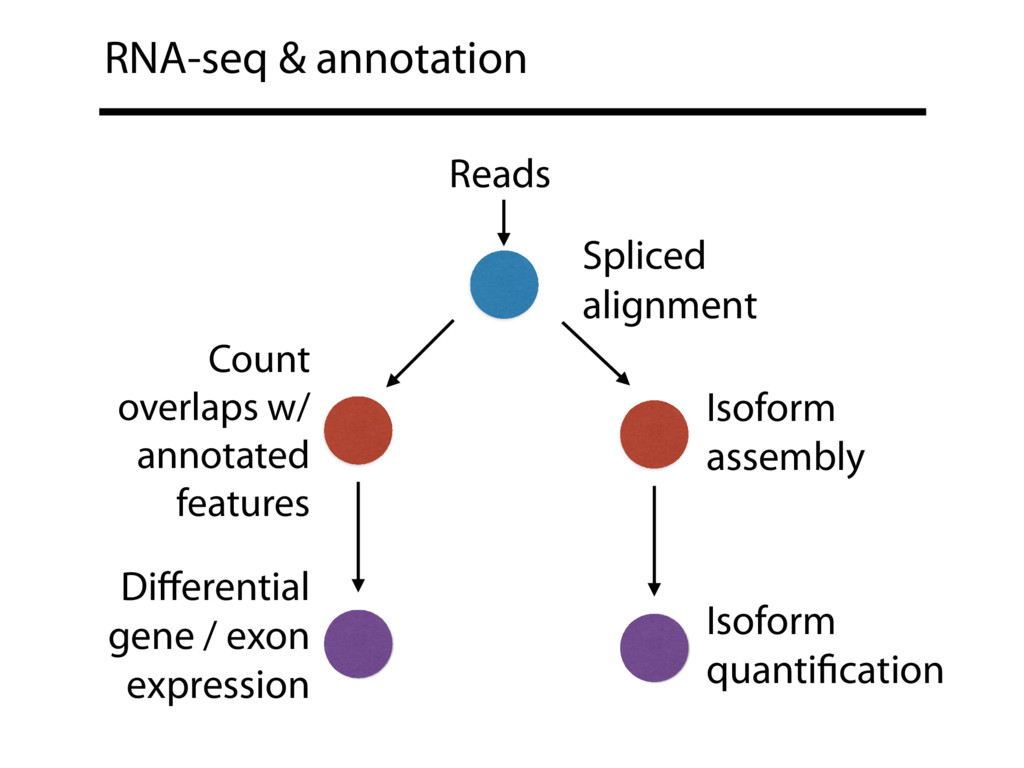{kind=link}
{kind=link}
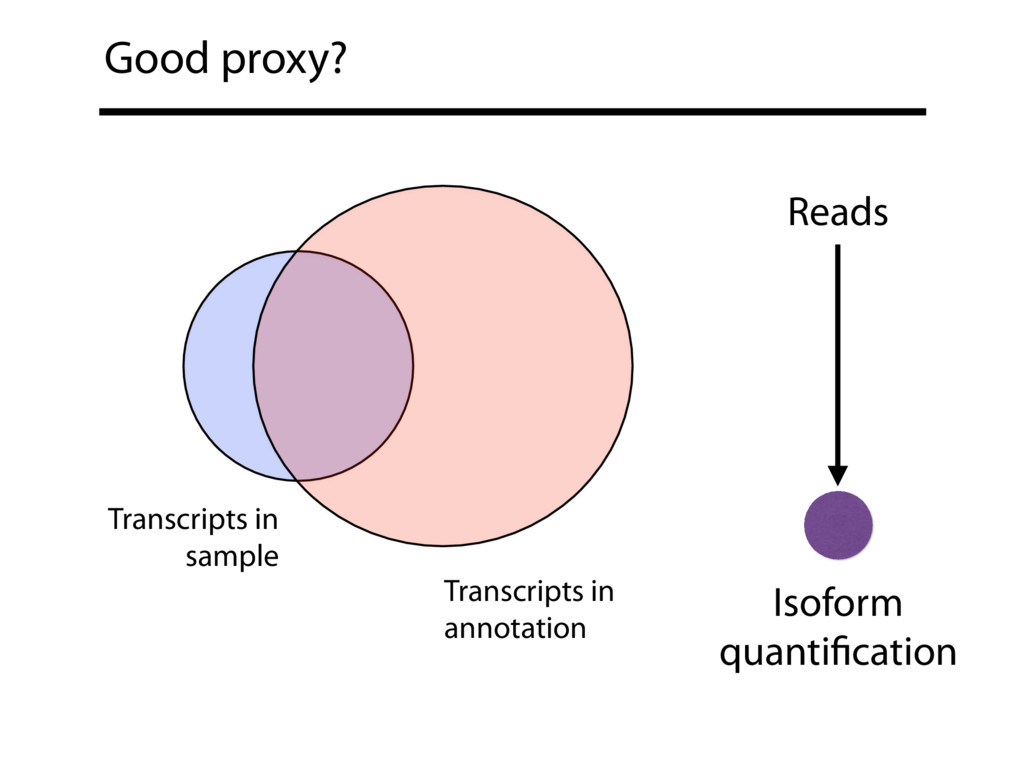{kind=link}
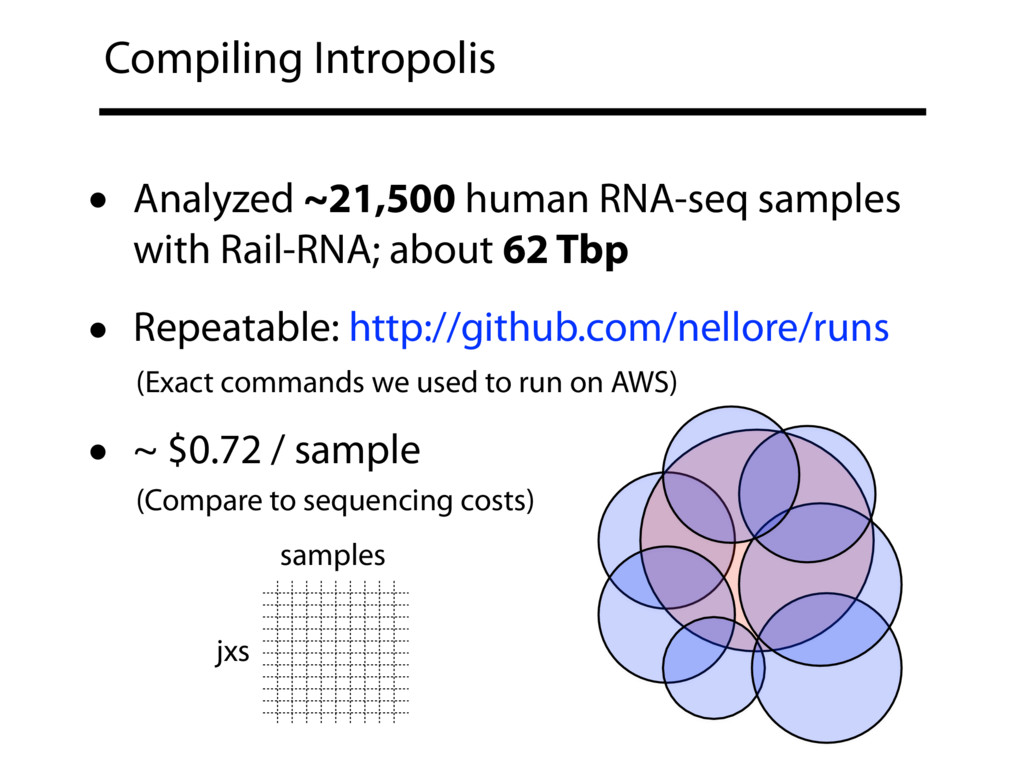{kind=link}
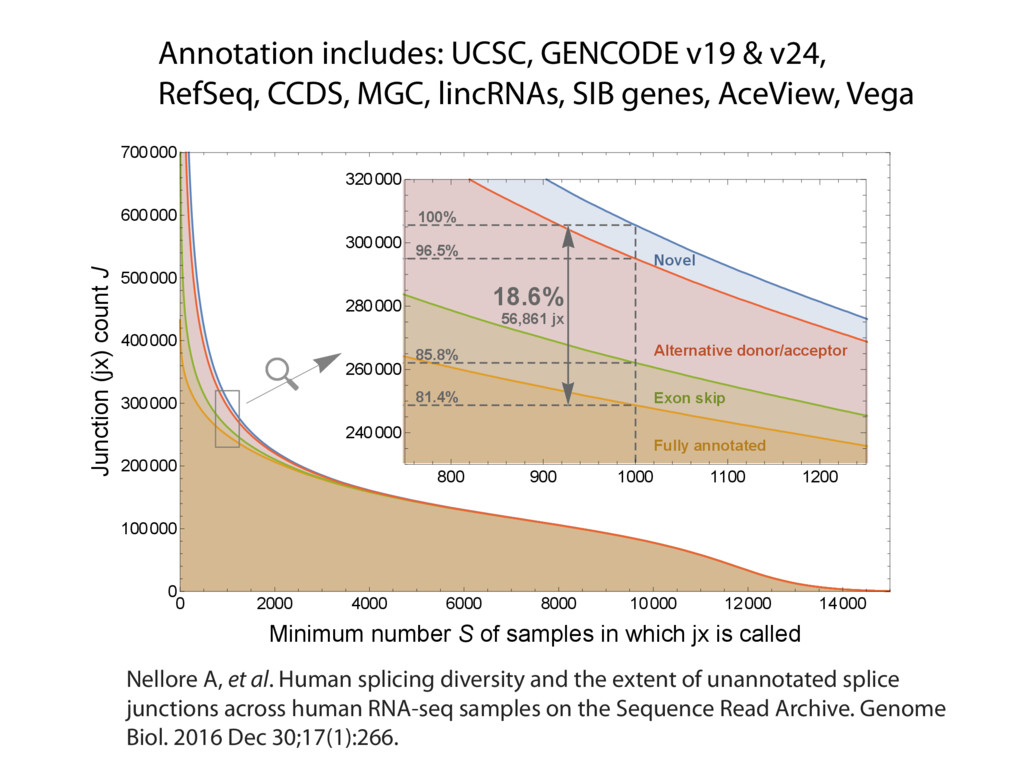{kind=link}
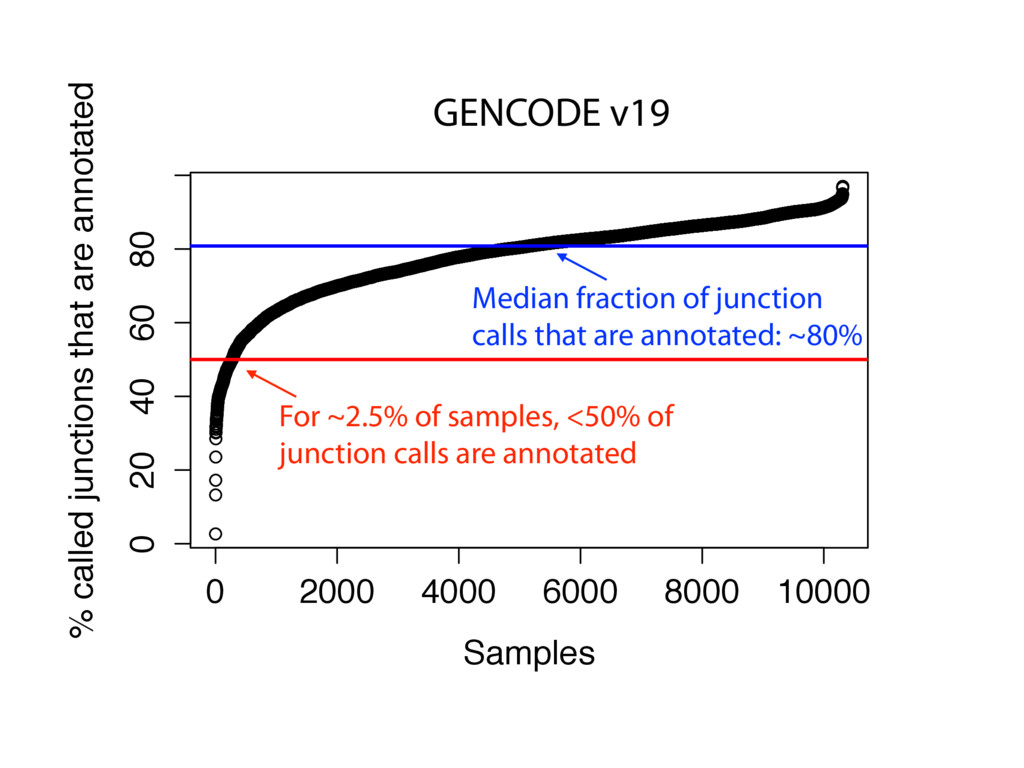{kind=link}
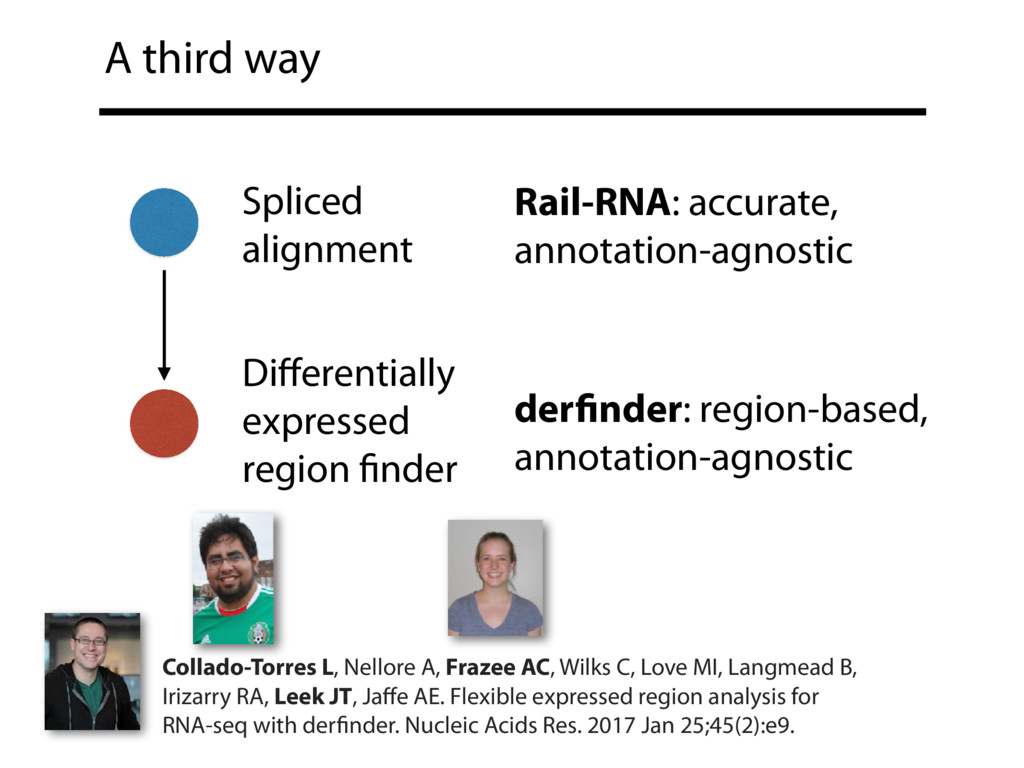{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}