solution. It is another TOOL at hand: • It is not cheap: Computationally expensive. Big molecules out of present implemen- tations. • At the moment, integration accuracy is sometimes hard to achieve. • It lies out of DFT. Perhaps with future implementations of density matrix functionals? • You have to be comfortable with QTAM. Some people are not. Then perhaps try your favorite partition method? • It provides magnitudes invariant under orbital transformations. You don’t have a partition into orbital contributions. Perspectives: • Use spin(full) ρ2 . • Use other thinner basins: ELF, ... • Build libraries of energies, chemical potentials, multipoles, and use them as molecular building blocks. • Examine systems. c ´ Angel Mart´ ın Pend´ as, 2005 (83) IQA Summary & Conclusions Bibliography: R. F. W. Bader, Atoms in Molecules. Oxford Univ. Press. Oxford 1990. L. Li and R. G. Parr, J. Chem. Phys. 84, 1704 (1986). J. Rychlewski and R. G. Parr, J. Chem. Phys. 84, 1696 (1986). K. Ruedenberg, Rev. Mod. Phys., 34, 326 (1962). F. M. Bickelhaupt and E. J. Baerends, Rev. Comput. Chem., 15, 1 (2000). G. Frenking et. al., Coord. Chem. Rev. 238-239, 55 (2003). S. Shaik et. al., J. Am. Chem. Soc. 114, 7861 (1992). A. Mart´ ın Pend´ as, Aurora Costales and V´ ıctor Lua˜ na. Phys. Rev. B 55, 4275 (1997). A. Mart´ ın Pend´ as, M. A. Blanco, and E. Francisco. J. Chem. Phys. 120, 4581 (2004). A. Mart´ ın Pend´ as, E. Francisco, and M. A. Blanco. J. Comput. Chem. 26, 344 (2005). M. A. Blanco, A. Mart´ ın Pend´ as, and E. Francisco, J. Chem. Phys., submitted. c ´ Angel Mart´ ın Pend´ as, 2005 (84)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Density AIM • Example: N2 CAS[10,10]//TZV(d,f) Re calculation. (Minimization of](https://files.speakerdeck.com/presentations/c58adbb069b9013170b6769b07168819/slide_19.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
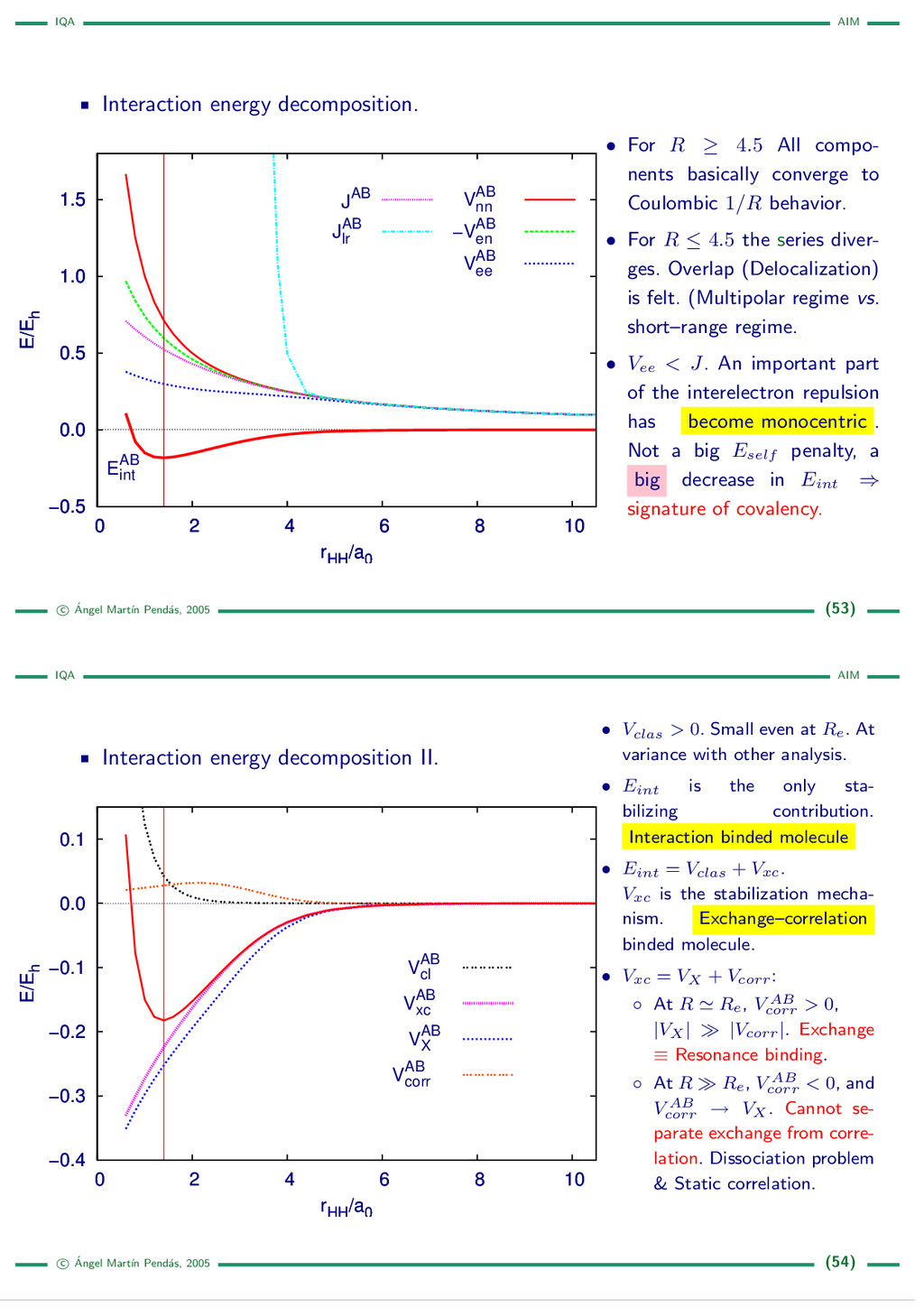{kind=link}
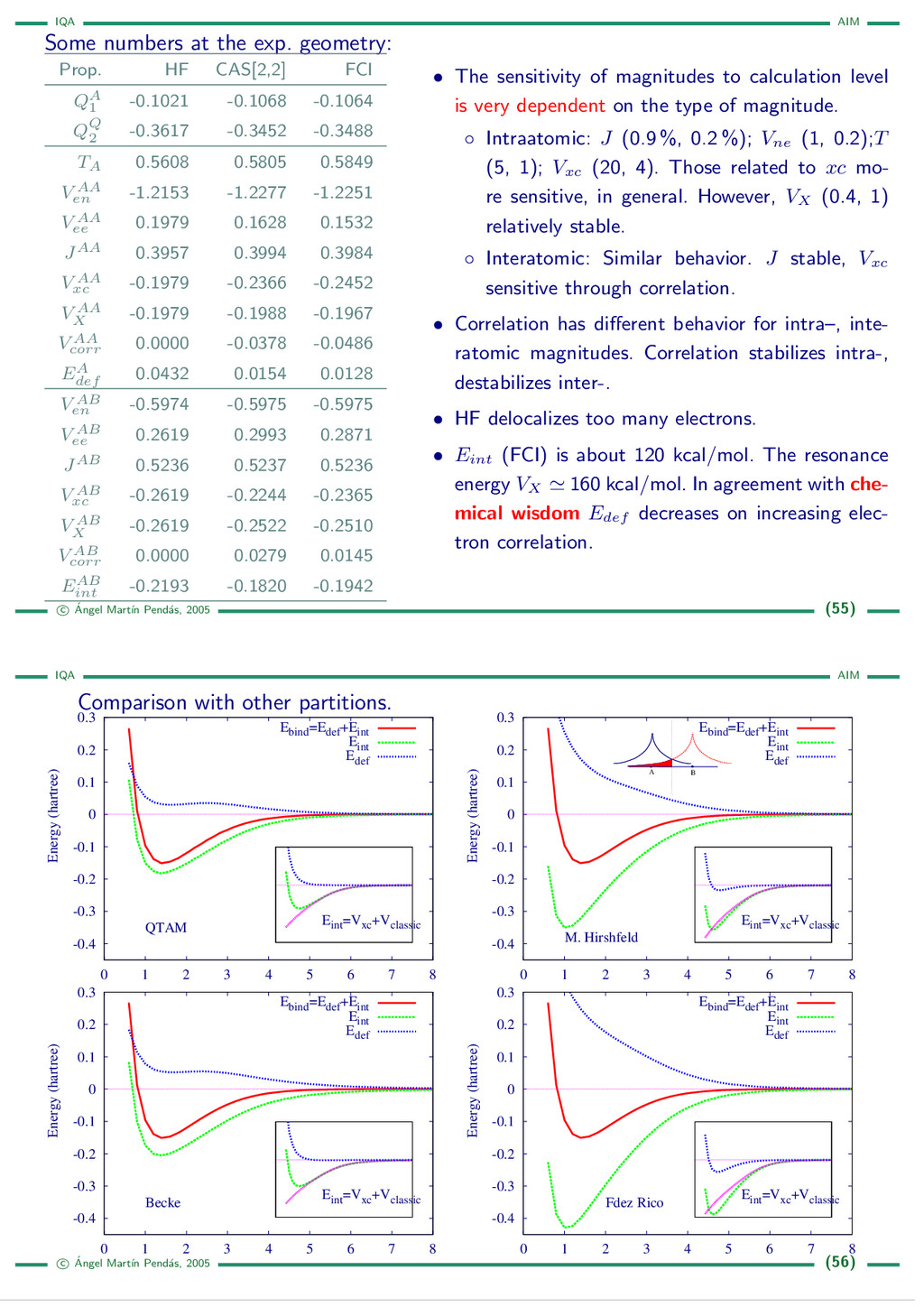{kind=link}
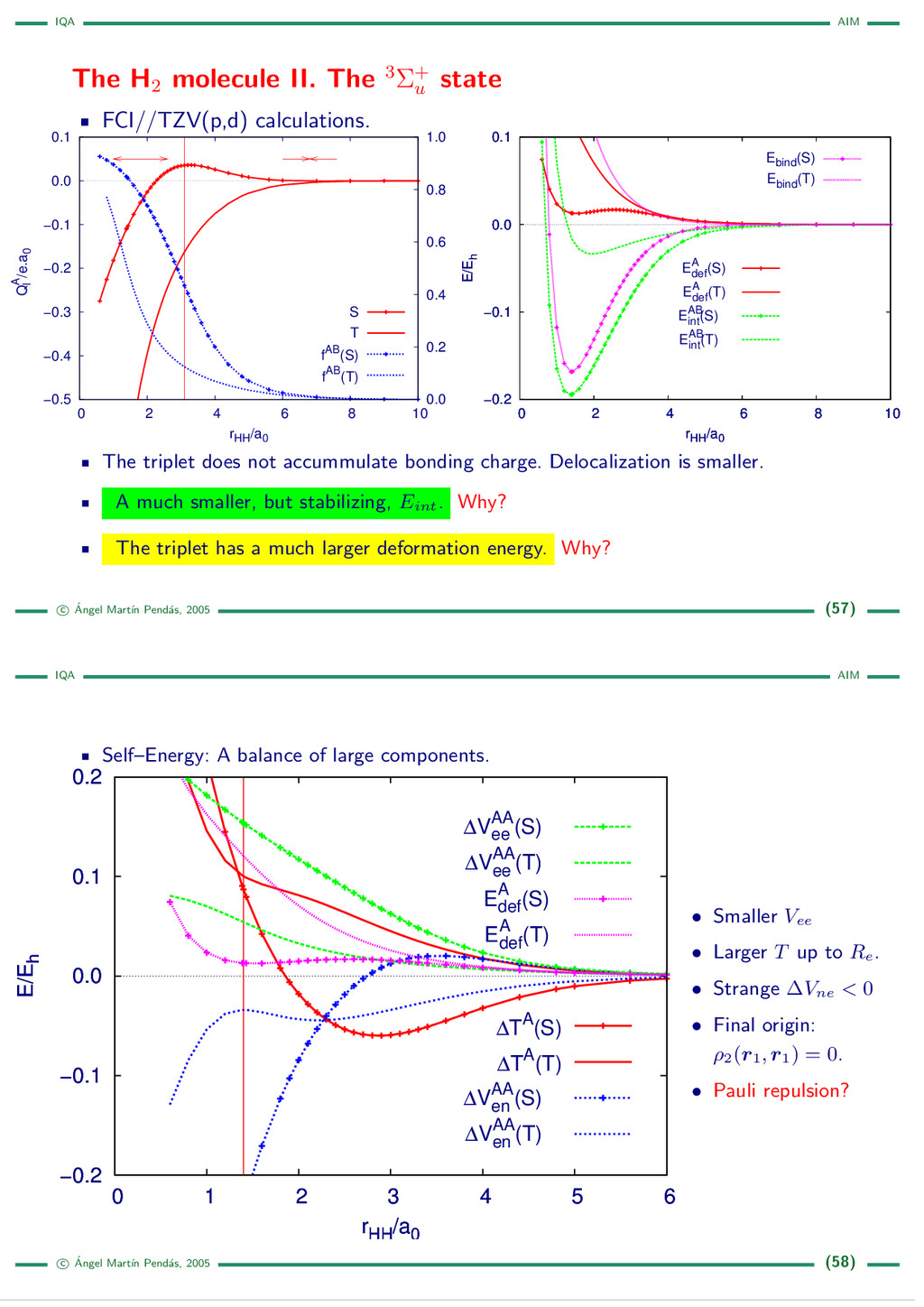{kind=link}
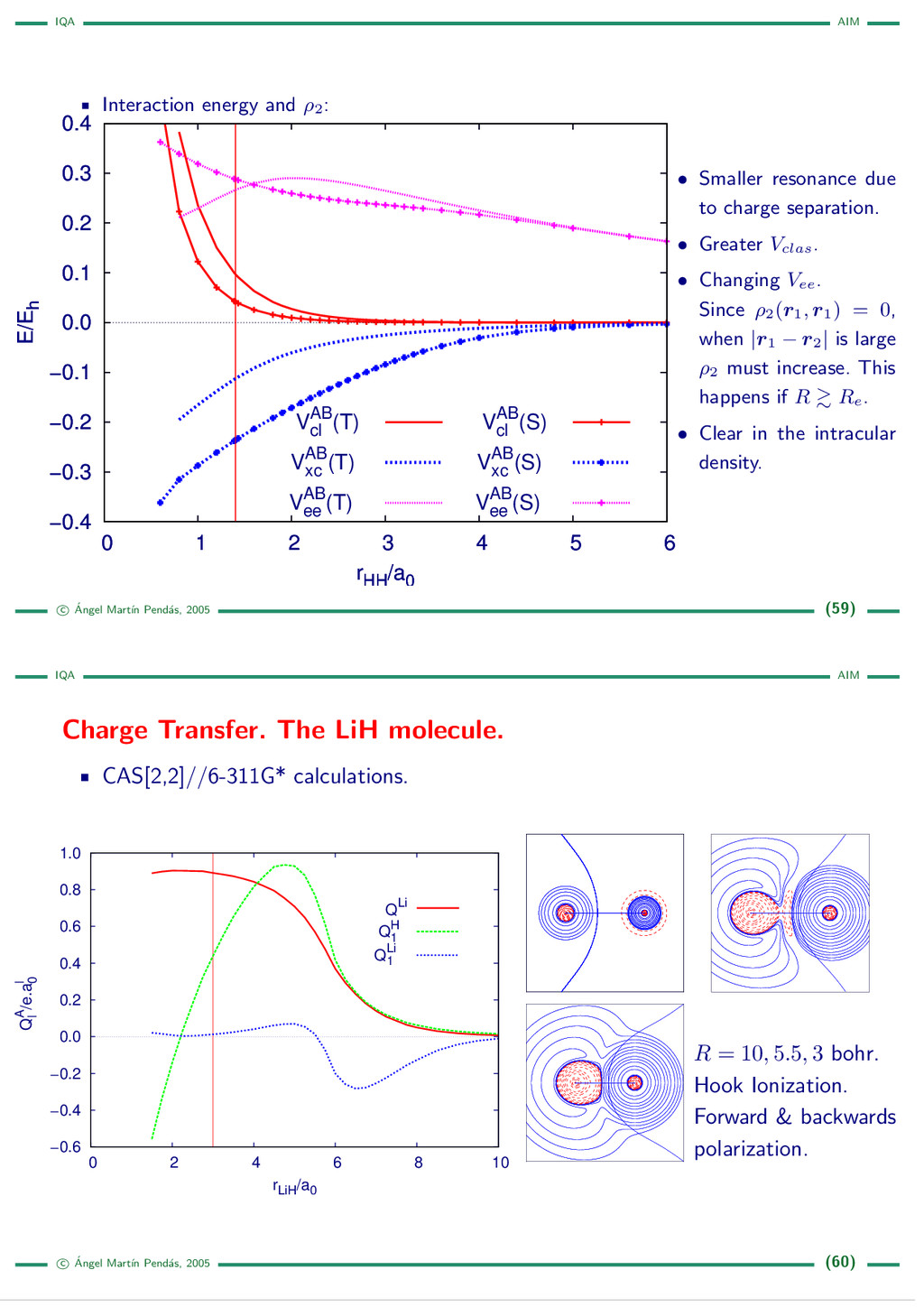{kind=link}
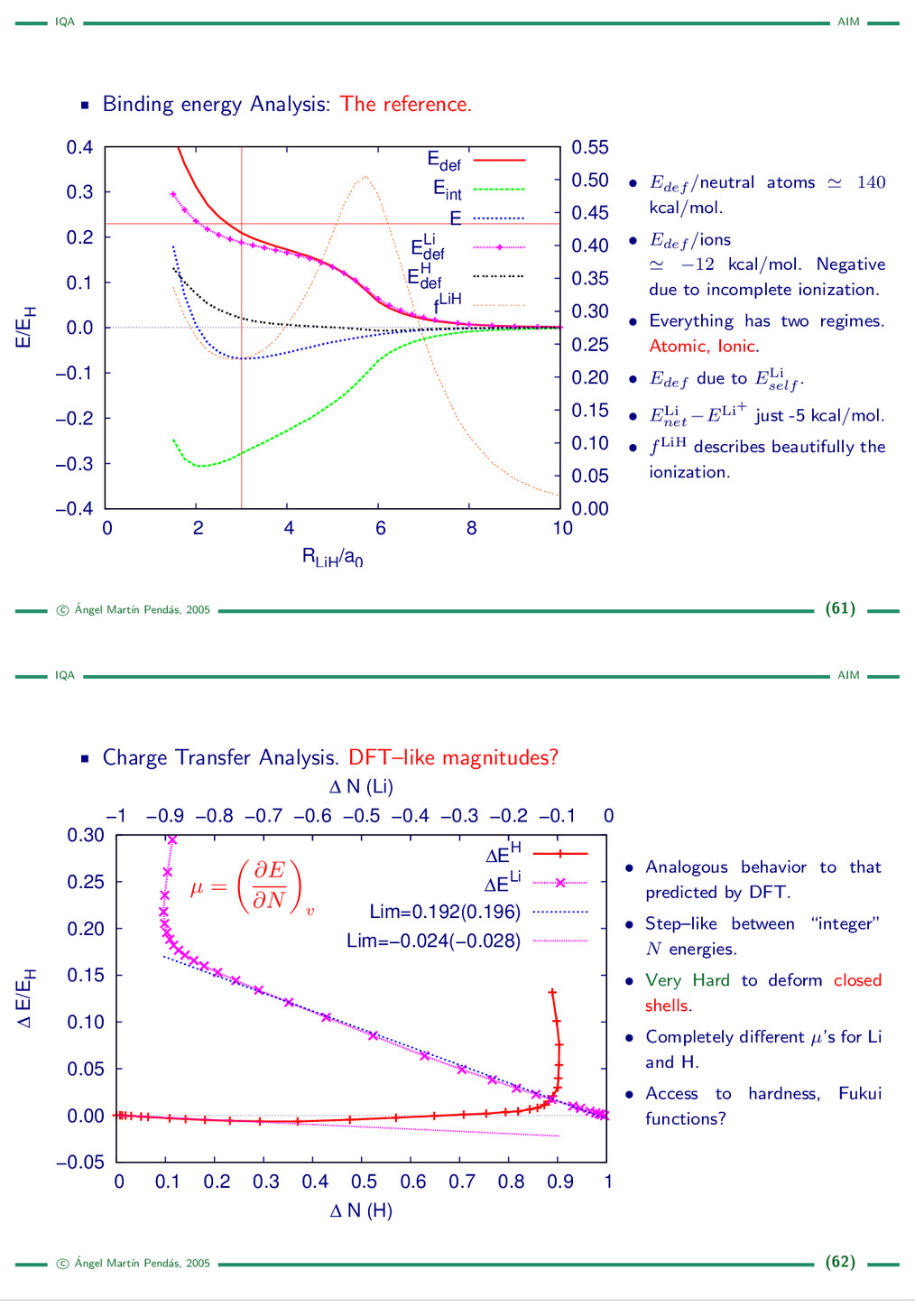{kind=link}
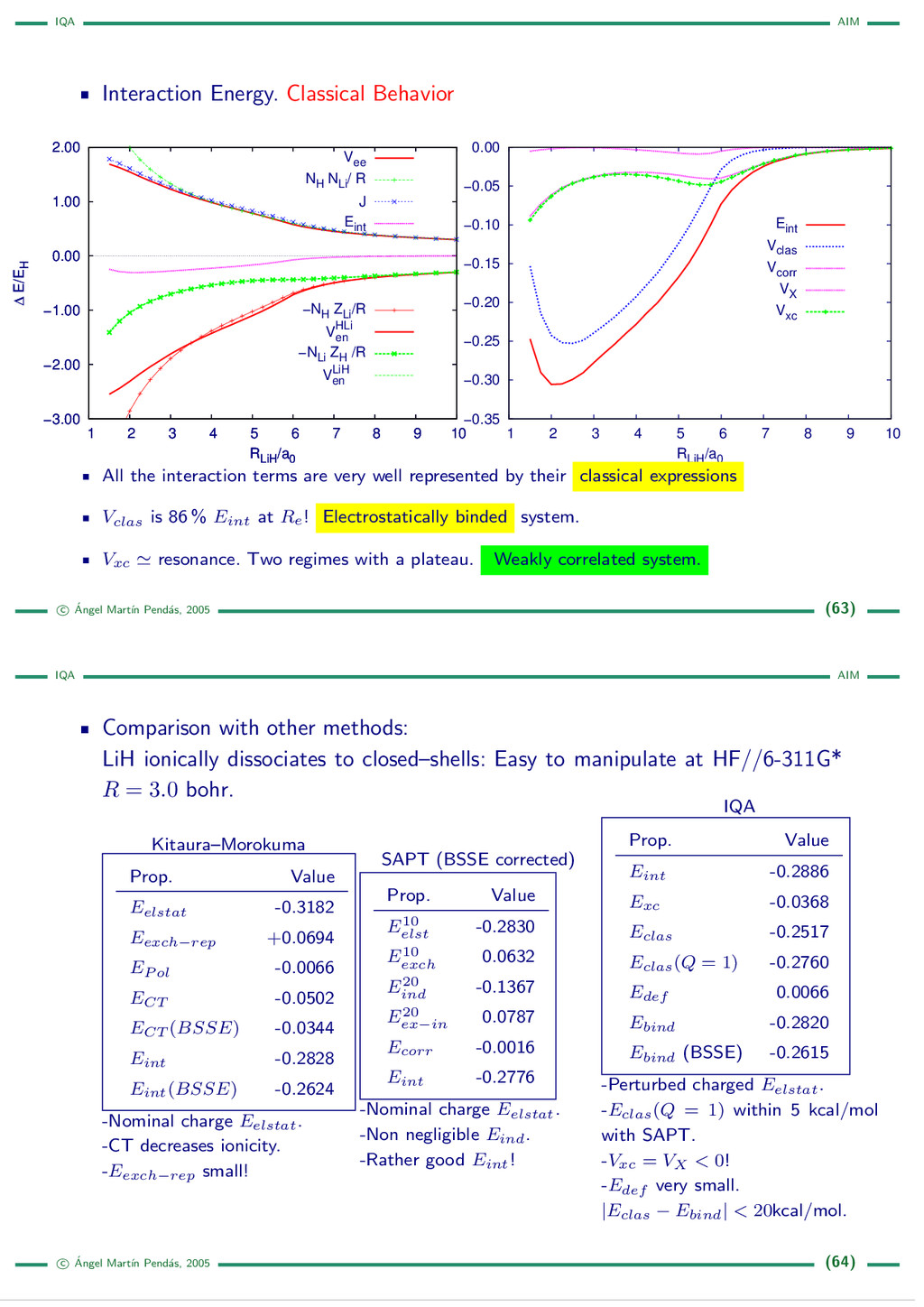{kind=link}
![IQA AIM Polar bonds: the 1A1 H2 O molecule. CAS[6,5]//6-311G**](https://files.speakerdeck.com/presentations/c58adbb069b9013170b6769b07168819/slide_32.jpg){kind=link}
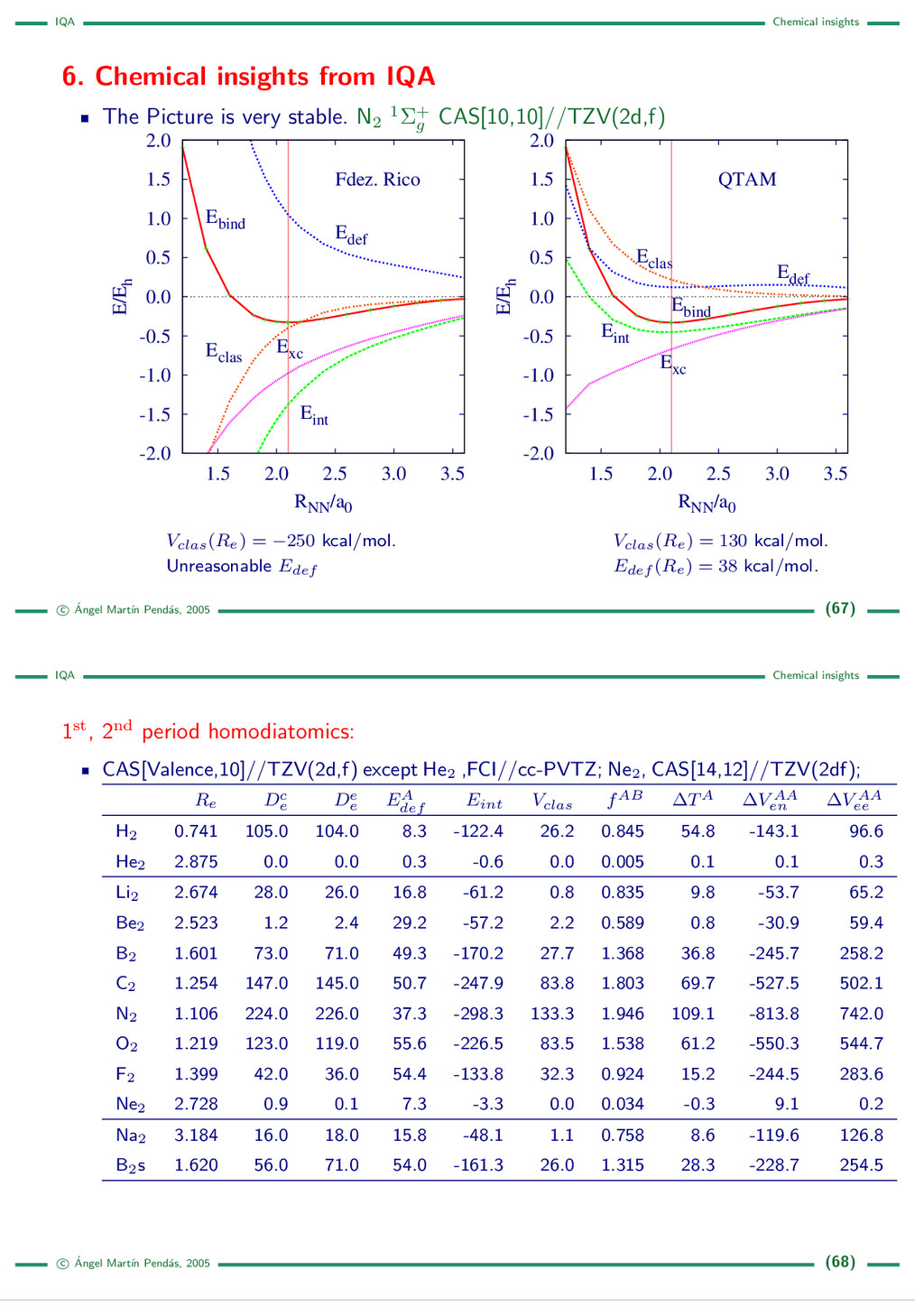{kind=link}
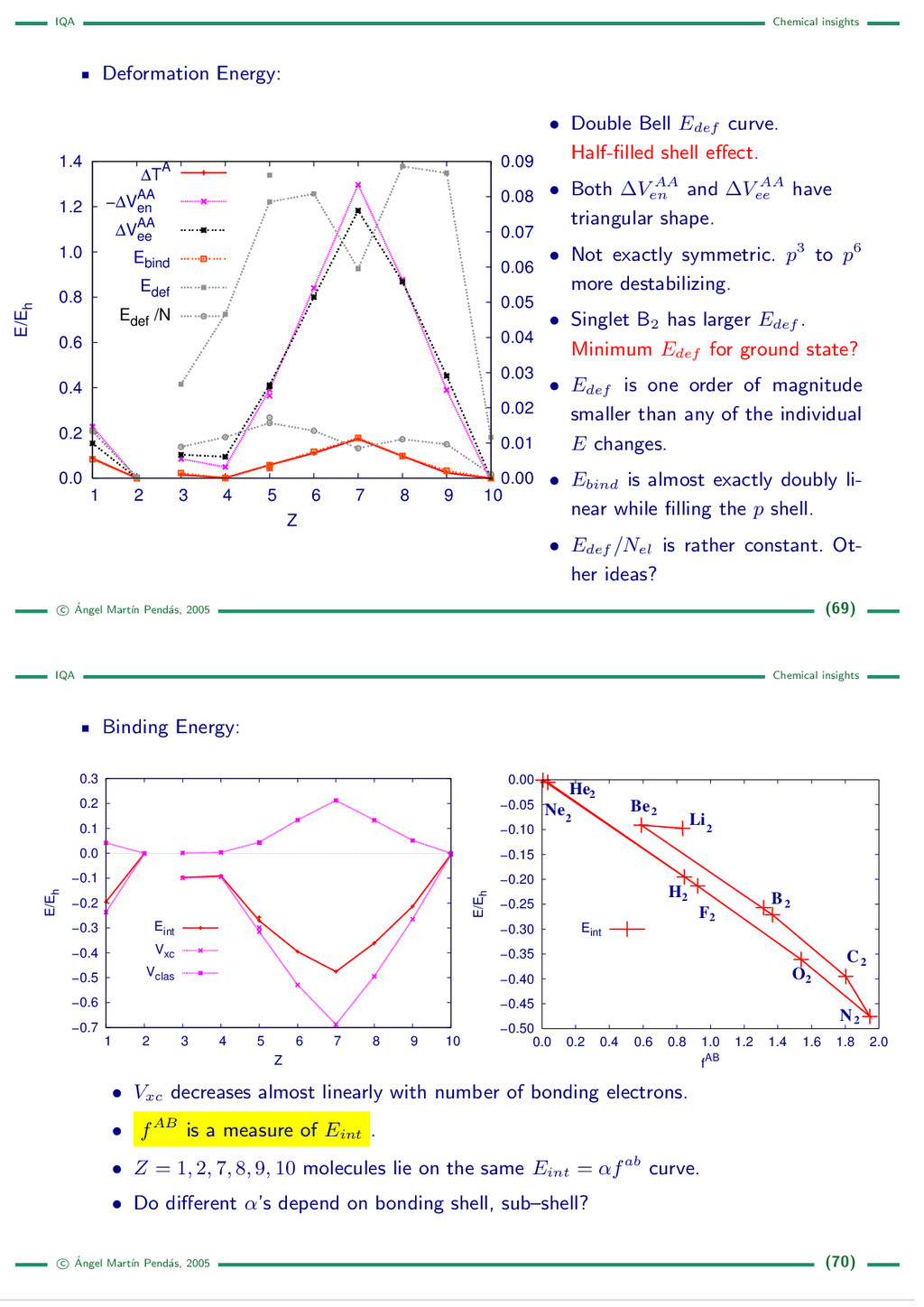{kind=link}
{kind=link}
![IQA Examples Bond formation and bond breaking. H2 O CAS[6,5]//6-311G**](https://files.speakerdeck.com/presentations/c58adbb069b9013170b6769b07168819/slide_36.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}