variables) that provide the most differentiating information between multiple phenotypes in my data set? – What are the OTUs that best differentiate between healthy and unhealthy patients? – Such knowledge is not only useful for classification, but also interpretation of a data sample • Feature selection can aid in the research of large sample data bases where examination of the data by hand is infeasible • We propose using a feature selection tool - based off of the Chi- squared test - for selection the top 15 features in a sample. – Deployment is on a KBase server for public use – Feature selection is implemented using the Scikits-Learn Python module
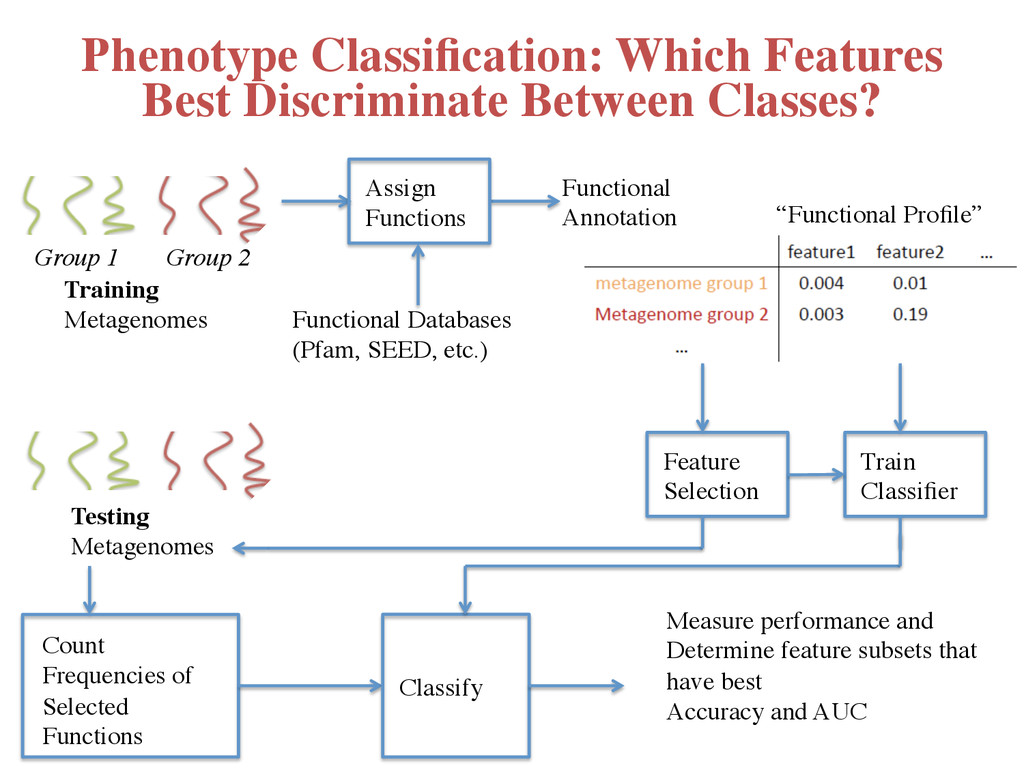
Profile” Feature Selection Count Frequencies of Selected Functions Training Metagenomes Group 1 Group 2 Testing Metagenomes Classify Train Classifier Measure performance and Determine feature subsets that have best Accuracy and AUC Phenotype Classification: Which Features Best Discriminate Between Classes?
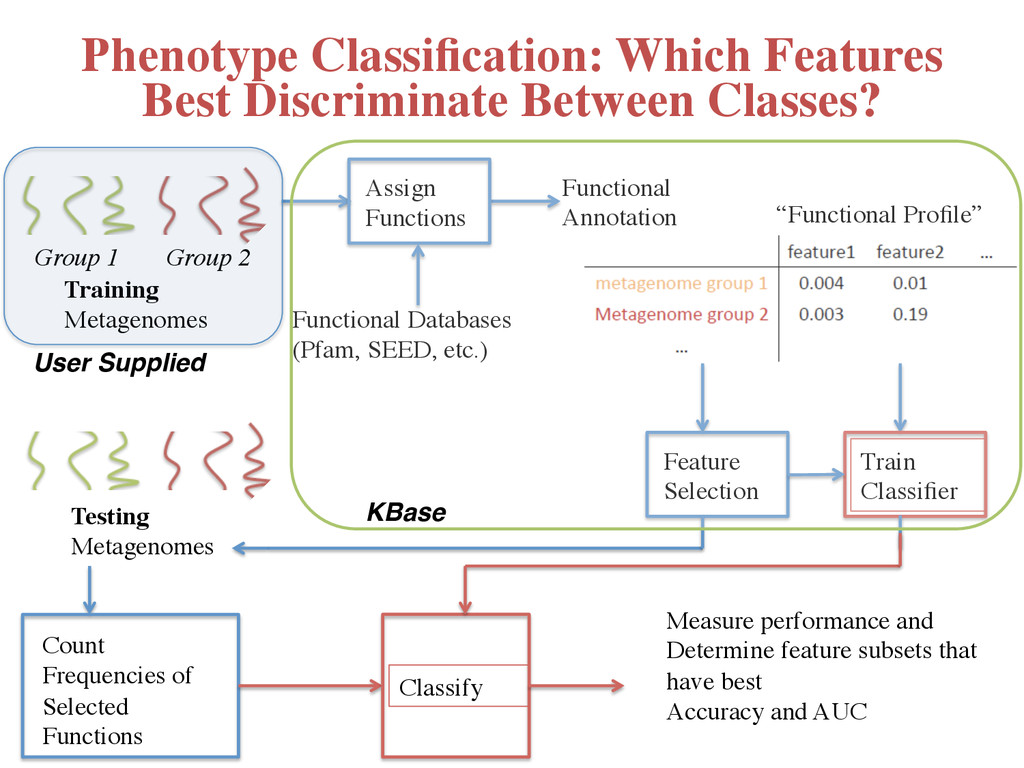
Profile” Feature Selection Count Frequencies of Selected Functions Training Metagenomes Group 1 Group 2 Testing Metagenomes Classify Train Classifier Measure performance and Determine feature subsets that have best Accuracy and AUC Phenotype Classification: Which Features Best Discriminate Between Classes? User Supplied! KBase!
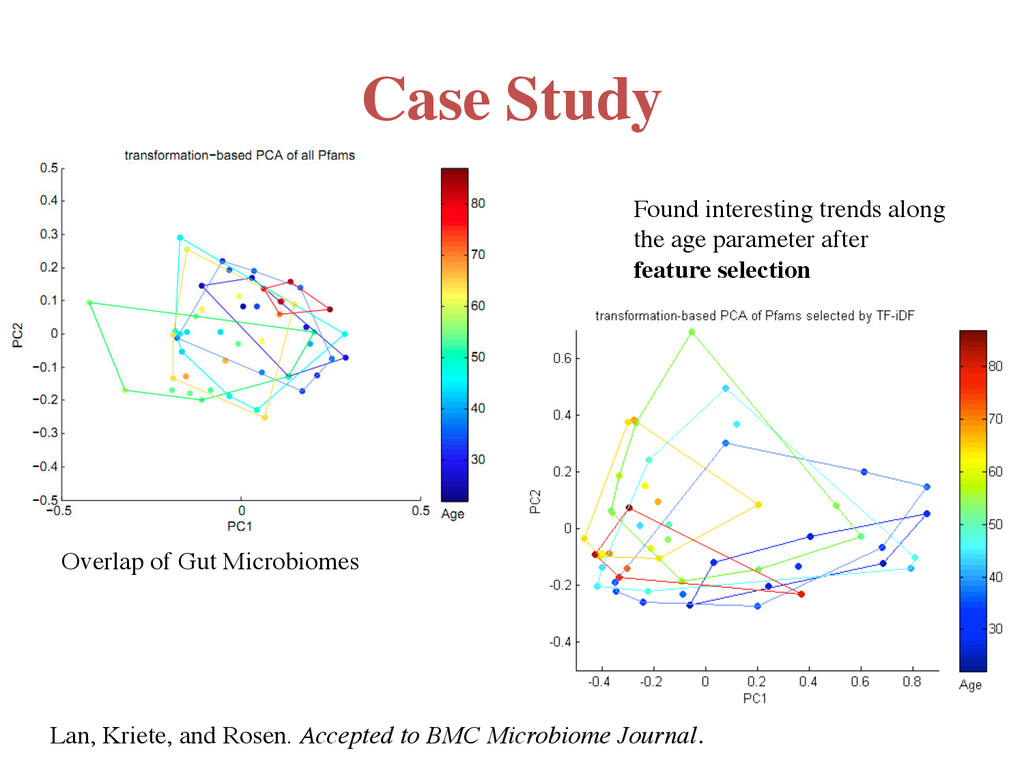
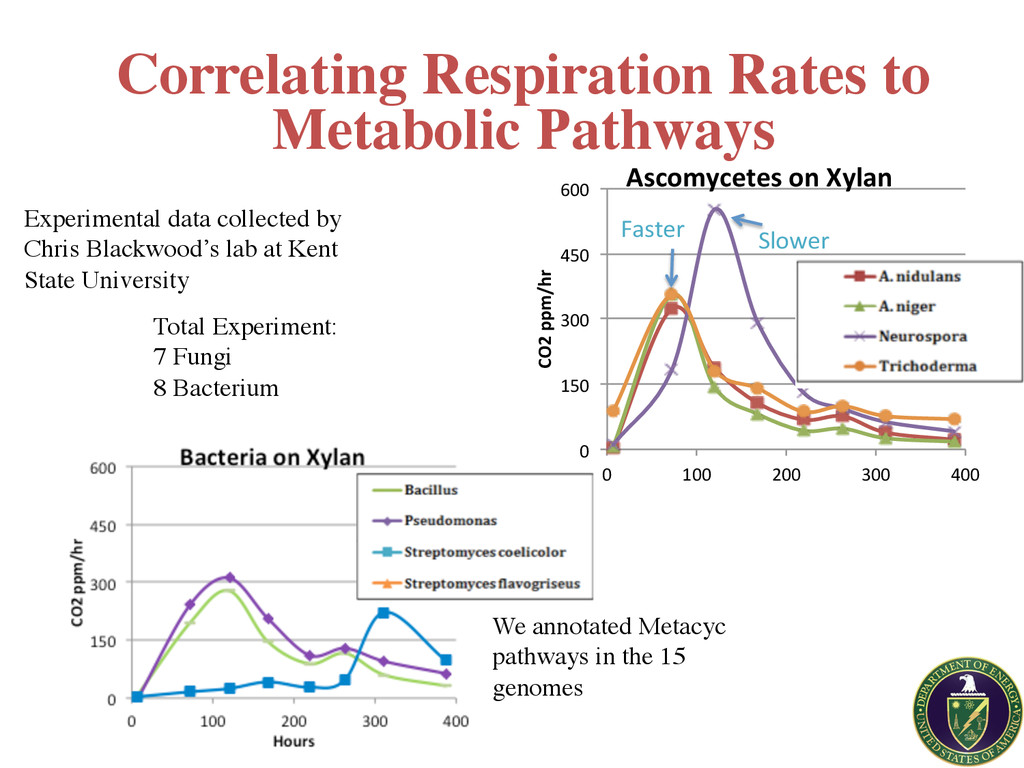
600" 0" 100" 200" 300" 400" CO2$ppm/hr$ Hours$ Ascomycetes$on$Xylan$ Faster Slower We annotated Metacyc pathways in the 15 genomes Experimental data collected by Chris Blackwood’s lab at Kent State University Correlating Respiration Rates to Metabolic Pathways
annotated for taxonomy and function • Implement a feature selection routine to identify relevant features in the learning problem • Users will be allowed to select from a variety of information theory methods for feature selection • Output: Users will be returned a list of relevant features
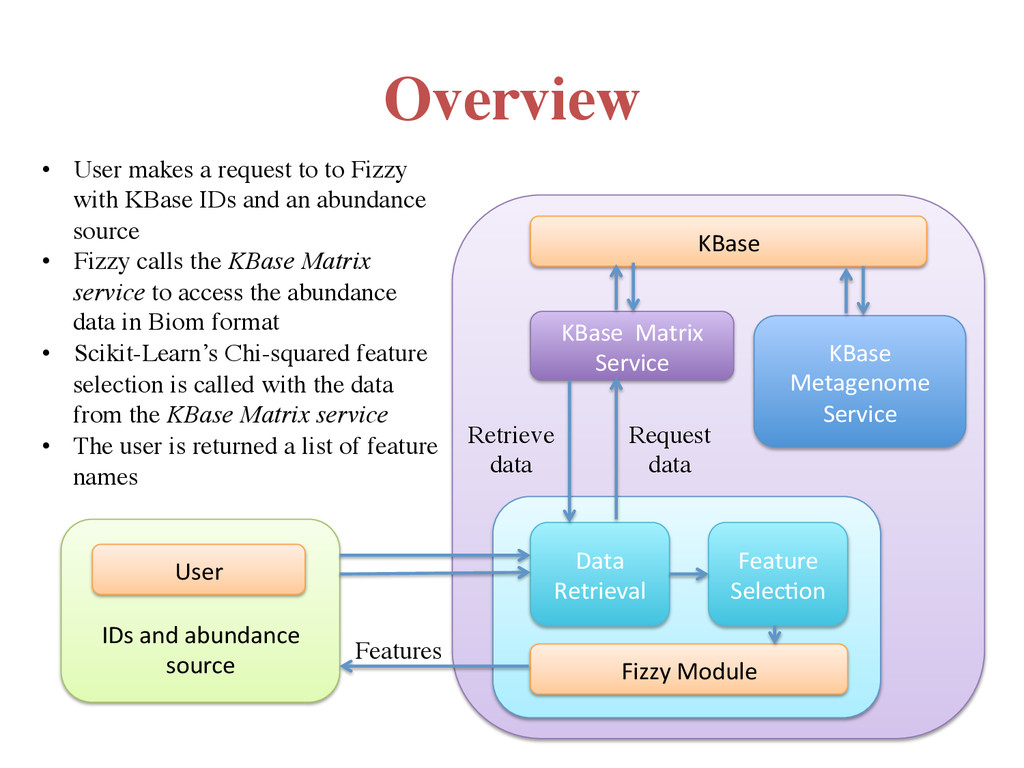
Service Feature Selec7on Data Retrieval User IDs and abundance source Request data Retrieve data Features KBase Metagenome Service • User makes a request to to Fizzy with KBase IDs and an abundance source • Fizzy calls the KBase Matrix service to access the abundance data in Biom format • Scikit-Learn’s Chi-squared feature selection is called with the data from the KBase Matrix service • The user is returned a list of feature names
dependence between random variable – Features and class labels are random variables – Features that are independent of the labels are non-informative for prediction and should be discarded • The feature selection method works by computing the Chi- squared statistic for each class and feature combination – Features are assumed to be independent – Top 15 scoring features are selected • Current implementation does not consider redundancy terms in the random variables
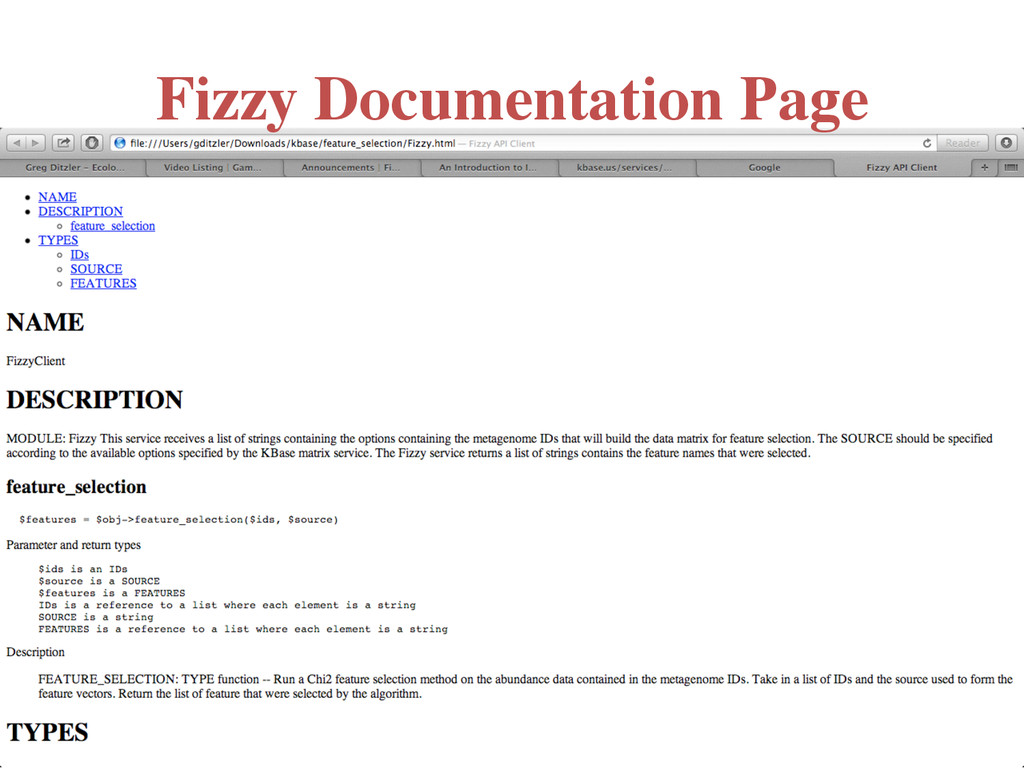
(list <string>) • Metagenome IDs that are currently available on KBase. The “label” information is assumed to be located in the metadata under: metadata::sample::data::biome • Future work will be extended to user uploaded data sets – Source (string) • M5NR, RefSeq, SwissProt, GenBank, IMG, SEED, TrEMBL, PATRIC, KEGG, M5RNA, RDP, Greengenes, LSU, and SSU – Authentication may be required if access to a “private” ID(s) is being requested • Program Output – List of feature names collected via feature selection. Any potential error may have been listed in the returned feature set. (list <string>)
selection – A Chi-squared test is one of many implementations of feature selection. We plan on integrating information theoretic feature selection methods, such as mRMR, into the KBase Fizzy module. – Efficient C-libraries exist for such implementations • Upload your own abundance tables – We are planning on extending Fizzy to use KBase services that allow users to upload their own data. In which case, we will work with an implementation of Fizzy that receives the data from the KBase upload module rather than metagenome IDs • Extended user control – Control over: feature selection method, the number of features to select, and parameters affiliated with feature selection
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
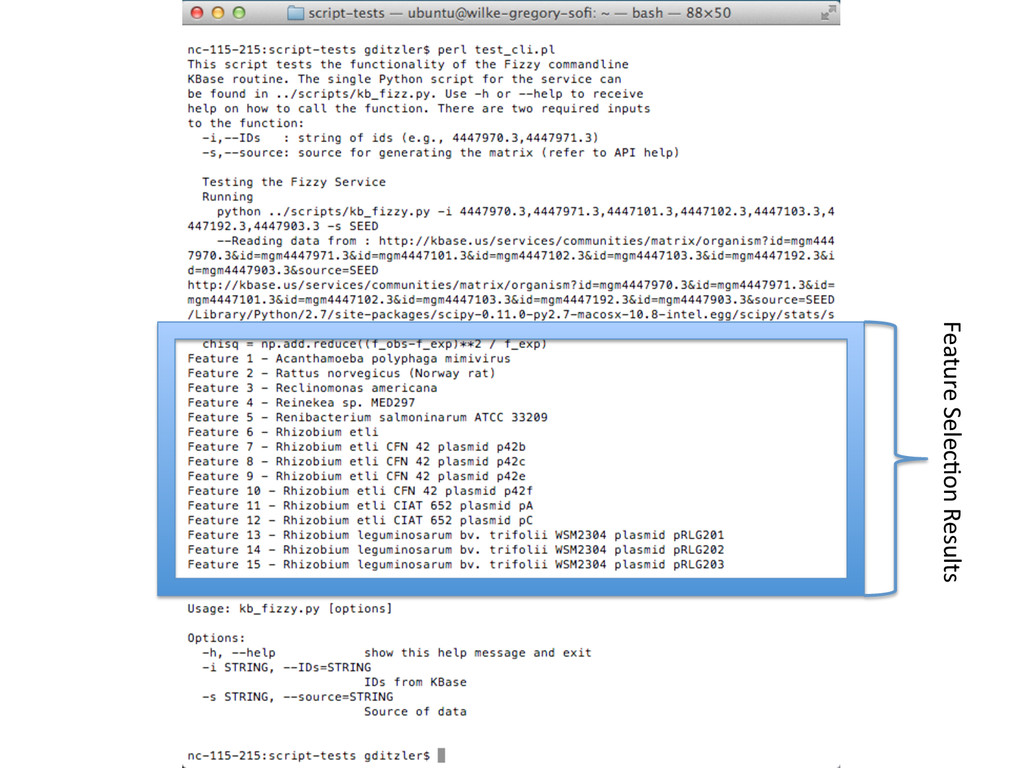
![Help with Fizzy (Python) gditzler$ ./kb_fizzy.py --help! Usage: kb_fizzy.py [options]!](https://files.speakerdeck.com/presentations/16e40340b24001315463327618870de4/slide_10.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}